
Post-translational S-Nitrosylation Is an Endogenous Factor Fine Tuning the Properties of Human S100A1 Protein*
- Martina Lenarčič Živković‡,1,
- Monika Zaręba-Kozioł§,1,
- Liliya Zhukova§,
- Jarosław Poznański§,
- Igor Zhukov‡§,2 and
- Aleksandra Wysłouch-Cieszyńska§,3
- From the ‡National Institute of Chemistry, Slovenian NMR Centre, Hajdrihova 19, 1001 Ljubljana, Slovenia and
- the §Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawińskiego 5a, 02-106 Warsaw, Poland
- ↵2 To whom correspondence may be addressed. Tel.: 48-22-592-2038; Fax: 48-22-592-2038; E-mail: igor{at}ibb.waw.pl.
- ↵3 To whom correspondence may be addressed. Tel.: 48-22-592-2340; Fax: 48-22-592-2340; E-mail: olawyslouch{at}ibb.waw.pl.
-
↵1 Both authors contributed equally to this work.
Abstract
S100A1 is a member of the Ca2+-binding S100 protein family. It is expressed in brain and heart tissue, where it plays a crucial role as a modulator of Ca2+ homeostasis, energy metabolism, neurotransmitter release, and contractile performance. Biological effects of S100A1 have been attributed to its direct interaction with a variety of target proteins. The (patho)physiological relevance of S100A1 makes it an important molecular target for future therapeutic intervention. S-Nitrosylation is a post-translational modification of proteins, which plays a role in cellular signal transduction under physiological and pathological conditions. In this study, we confirmed that S100A1 protein is endogenously modified by Cys85 S-nitrosylation in PC12 cells, which are a well established model system for studying S100A1 function. We used isothermal calorimetry to show that S-nitrosylation facilitates the formation of Ca2+-loaded S100A1 at physiological ionic strength conditions. To establish the unique influence of the S-nitroso group, our study describes high resolution three-dimensional structures of human apo-S100A1 protein with the Cys85 thiol group in reduced and S-nitrosylated states. Solution structures of the proteins are based on NMR data obtained at physiological ionic strength. Comparative analysis shows that S-nitrosylation fine tunes the overall architecture of S100A1 protein. Although the typical S100 protein intersubunit four-helix bundle is conserved upon S-nitrosylation, the conformation of S100A1 protein is reorganized at the sites most important for target recognition (i.e. the C-terminal helix and the linker connecting two EF-hand domains). In summary, this study discloses cysteine S-nitrosylation as a new factor responsible for increasing functional diversity of S100A1 and helps explain the role of S100A1 as a Ca2+ signal transmitter sensitive to NO/redox equilibrium within cells.
Introduction
S100A1 is a prominent example of the S100 family of small EF-hand type Ca2+-binding proteins. The protein is an important factor in Ca2+ signaling pathways in a variety of cell types, including neuronal, cardiac, and vascular cells (1, 2). It plays multifaceted roles in human physiology via Ca2+-dependent and -independent binding to target proteins (3, 4). Through binding with multiple intracellular and extracellular targets, S100A1 is involved in the regulation of neurotransmitter release in the brain (5), cardiomyocyte Ca2+ cycling (2, 6), contractile performance, and energy balance in the heart (7, 8). S100A1 is a marker of several human diseases (9) and a target molecule for therapy of heart failure (10, 11). Methods of increasing the level of S100A1 protein are being studied as important therapeutic strategies to improve heart contractility in heart failure, which are free of the severe side effects caused by clinically used inotropic agents, such as β-blockers (12). The outcome of restoring S100A1 levels in models of failing heart is different with the protein delivered exogenously than with the one expressed following a viral gene transfer (13). From this point of view, understanding the factors regulating human S100A1 function is of special importance.
Post-translational modifications are a ubiquitous way of increasing the functional diversity of proteins. S100A1 belongs to a subclass of S100 proteins that possess a conserved cysteine residue in the C-terminal fragment of the protein (supplemental Fig. S1). As we have shown previously, the C-terminal cysteine thiol group is hyperreactive toward nitric oxide donors for at least two members of this S100 protein subgroup. Reaction with an important intracellular nitric oxide donor, S-nitrosoglutathione, leads to S-nitrosylation of the C-terminal cysteine in recombinant S100A1 and S100B proteins (14). Experiments on recombinant proteins have shown that S-nitrosylation modifies the interaction of S100B protein with its important target p53 (15). Moreover, studies on chemically modified recombinant S100A1 protein have shown that several covalent modifications of the very reactive Cys85 (S-glutathionylation (16), alkylation (17), or formation of mixed disulfides with β-mercaptoethanol (18)) result in an increase of Ca2+ affinity to both EF-hand loops. On the other hand, in vitro experiments of Ca2+ binding to S100A1 protein show a significant decrease in the binding affinity in buffers with close to physiological ionic strength in comparison with low ionic strength solutions (19).
Cysteine thiol group modifications, in particular S-nitrosylation, play a crucial role in the brain and cardiac signal transduction under physiological and pathological conditions (20,–,22). So far, the regulatory mechanism of protein S-nitrosylation has not been fully characterized. There are no reliable ways to predict the site of S-nitrosylation in a protein, and until now only experimental methods have been used to detect this modification.
In this work, we demonstrate that S100A1 is endogenously S-nitrosylated in cells. To prove that, we used the PC12 model cell line, which is frequently used in the studies of S100A1 activity (23). We have studied the effects of S-nitrosylation on Ca2+ binding and on the high resolution three-dimensional structure of human apo-S100A1,4 aiming at the elucidation of yet unknown factors regulating various biological functions of the protein. For this purpose, stable chemically S-nitrosylated recombinant human S100A1 protein was used. In contrast with previously published structural studies on S100A1, the experiments presented in this work were performed at physiological ionic strength and pH. We have determined the in-solution, high resolution three-dimensional structure of apo-S100A1 protein with S-nitrosylated C-terminal Cys85 (apo-S100A1-NO) and compared it with that of unmodified apo-S100A1 protein. The results suggest that S-nitrosylation might regulate the biological activity not only of S100A1 itself but also of other members of the subclass of S100 proteins that have a C-terminal cysteine in the sequence.
EXPERIMENTAL PROCEDURES
Expression and Purification of Recombinant S100A1 Protein
The synthetic gene encoding for human S100A1 was cloned into pET-30a+ plasmid and expressed in Escherichia coli utilizing the T7 expression system. Bacterial cells were grown in LB medium at 37 °C. Expression was induced by the addition of 0.4 mm isopropyl 1-thio-β-d-galactopyranoside at A600 = 0.8, and the bacterial culture was grown for 2 h. The overexpressed protein was purified as described previously (24). Protein concentration was estimated from its absorbance at 280 nm. The molar extinction coefficient for S100A1 protein (ϵmolar = 10,026) was determined experimentally by measuring the UV signal at 280 nm for a standard S100A1 solution. Concentration of the standard S100A1 sample was established by amino acid analysis at BioCentrum Ltd. (Kraków, Poland). Namely, the protein samples were hydrolyzed in gas phase using 6 m HCl at 115 °C for 24 h. The liberated amino acids were converted into phenylthiocarbamyl derivatives and analyzed by high pressure liquid chromatography (HPLC) on a PicoTag 3.9 × 150-mm column (Waters, Milford, MA).
Transnitrosylation of S100A1 Protein with S-Nitrosoglutathione
S-Nitrosoglutathione (GSNO) was prepared by mixing 220 μl each of GSH in water and sodium nitrite in 0.1% TFA. The solution was incubated in the dark, under nitrogen for 10 min (final concentration of ∼100 mm GSNO) and used immediately after preparation. The final concentration of the GSNO stock solution was calculated from its absorbance at 334 nm. S-Nitrosylation of 13C,15N-double-labeled and unlabeled S100A1 was performed under denaturing conditions as published previously (14). 0.5 mg of lyophilized, purified, reduced S100A1 was dissolved in 100 μl of 6 m guanidinium hydrochloride, 20 mm Tris-HCl (pH 8.0). After the addition of 20 μl of freshly prepared GSNO (100 mm), the reaction was kept for 10 min in the dark. The reaction mixture was then diluted 6 times with water, and 100 μl of EDTA (0.25 m, pH 8.0) was added. The proteins were purified using analytical HPLC (80% yield after purification). Lyophilized proteins were refolded and used for further experiments. CD spectra for both apo-S100A1 and apo-S100A1-NO were almost identical and were typical for highly helical proteins.
Cell Culture Experiments
PC12 pheochromocytoma cells were a kind gift of Prof. Jacek Kuźnicki (The International Institute of Molecular and Cell Biology, Warsaw, Poland). The cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2, in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin (Invitrogen), and 100 units/ml streptomycin (Invitrogen). Before further analysis, intact cells were washed with PBS and cultured in DMEM supplemented with 1% FBS for 24 h. Confluent cells were scraped and resuspended in 250 mm HEPES buffer (pH 7.7) containing 1 mm EDTA and 0.1 mm neocuproine (HEN buffer). Cells were homogenized using a Potter glass homogenizer and centrifuged, and the lysate was used for further experiments.
Enrichment of S-Nitrosylated Proteins from Protein Lysates Using the Biotin Switch Method (BSM)
Total protein concentration of the PC12 lysates was measured using the Bradford test and adjusted to 1 mg/ml. Free thiols of the lysate proteins were S-methylated using 20 mm methyl methanethiosulfonate (Sigma) in 250 mm HEN buffer with 5% SDS (HENS buffer) at 50 °C for 20 min with agitation. After thiol blocking, proteins were precipitated with acetone and resuspended in the same volume of HEN buffer with 2.5% SDS. The protein solution was divided into two equal parts. One part was treated simultaneously with freshly prepared sodium ascorbate (final concentration 5 mm) and biotin-HPDP (Pierce; final concentration 400 μm). The second part was treated only with the biotin-HPDP solution devoid of the ascorbate. All samples were incubated in the dark for 1 h at room temperature. The proteins were precipitated; resuspended in the same volume of HENS buffer, diluted with 2 volumes of 20 mm Hepes buffer (pH 7.7), 100 mm NaCl, 1 mm EDTA; and incubated for 1 h at room temperature with 50 μl of neutravidin-agarose (Pierce). Afterward, the affinity resin beads were washed five times with high salt buffer (20 mm Hepes buffer, pH 7.7, 600 mm NaCl, 1 mm EDTA) and incubated for 20 min at room temperature with a 50 mm DTT solution in 50 mm Tris-HCl (pH 8.0), containing 1 mm EDTA. Fractions were collected and analyzed using protein detection on 4–20% SDS-Tricine polyacrylamide gels. For additional control experiments, recombinant S100B-NO nitrosylated solely at Cys84 was prepared as described elsewhere (14). S100B-NO solutions were treated with exactly the same buffers and biotin switch method as proteins from PC12 cells to prove the efficiency of the procedure. The presence of S100B in appropriate fractions after BSM was confirmed by Western blot using goat anti-S100B polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA).
Detection of S100A1 Protein in the Fraction of S-Nitrosylated Proteins from PC12 Pheochromocytoma Cells
The fraction of proteins enriched by the biotin switch method from whole cell lysate of PC12 pheochromocytoma cells was separated using 4–20% SDS-Tricine polyacrylamide gels and transferred to PVDF membranes (0.22 μm). The PVDF membrane was blocked with nonfat dried milk and incubated with diluted 1:2000 goat anti-S100A1 polyclonal antibody (Santa Cruz Biotechnology, Inc.) for 1 h and with 1:10000 diluted rabbit anti-goat HRP antibody for 1 h. S100A1 protein bands were detected using the ECL chemiluminescence system (Amersham Biosciences).
Isothermal Titration Calorimetry (ITC)
Calorimetric experiments for Ca2+ binding to apo-S100A1 and apo-S100A1-NO were monitored using the Nano ITC microcalorimeter (TA Instruments). Experiments
were performed at 25 °C. Protein samples and titrants contained 10 mm TES buffer, pH 7.2. 150 mm NaCl was present in both sample and titrant solutions. Prior to running the experiments, all solutions were degassed and
thermostatted at 25 °C. 5 mm CaCl2 solutions were injected in volumes of 5 μl in a series of 62 controlled doses separated by 400-s equilibration delays. The
final concentration of CaCl2 in the sample cell of 0.95-ml volume was close to 1 mm. Protein concentrations were in the range from 40 to 100 μm (subunit concentration), depending on the experiment. Heat flow data were interpreted by using a model that assumes sequential
binding of Ca2+ to three different types of binding sites, with the third type of the occupancy equal to n, as described by the following set of Reactions 1–3.
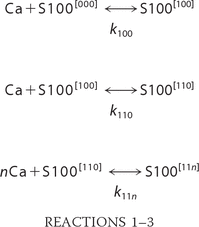 Accordingly, concentrations of all species present in solution satisfy the following equations.
Accordingly, concentrations of all species present in solution satisfy the following equations.
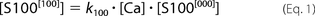
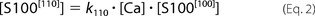
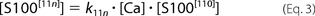
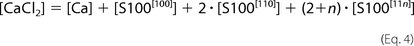
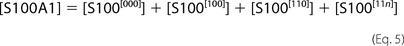 The [ijk] index describes appropriate occupancy of the three types of Ca2+ binding sites. [CaCl2] and [S100A1] are total concentrations of calcium chloride and S100A1 protein in the ITC sample cell. The ensemble of five
equations with five unknowns ([Ca], S100[000], S100[100], S100[110], and S100[11n]) was solved against the concentration of free protein, S100A1[000]. The obtained quartic function was resolved explicitly using a symbolic calculation module implemented in Mathematica version
8.0 (25). Experimentally measured incremental heat response upon ith injection, ΔHexp(i), was then modeled as a function of reagent concentrations according to the formula,
The [ijk] index describes appropriate occupancy of the three types of Ca2+ binding sites. [CaCl2] and [S100A1] are total concentrations of calcium chloride and S100A1 protein in the ITC sample cell. The ensemble of five
equations with five unknowns ([Ca], S100[000], S100[100], S100[110], and S100[11n]) was solved against the concentration of free protein, S100A1[000]. The obtained quartic function was resolved explicitly using a symbolic calculation module implemented in Mathematica version
8.0 (25). Experimentally measured incremental heat response upon ith injection, ΔHexp(i), was then modeled as a function of reagent concentrations according to the formula,
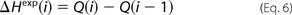
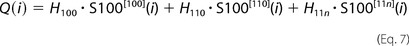 where Q(i) is the estimated heat release after i succeeding injections, and S100[ijk](i) are populations of variously Ca2+-loaded S100A1 forms after the ith injection, calculated according to the equations above. The values of stability constants (k100, k110, and k11n) and accompanying heats of binding (H100, H110, and H11n) were optimized to obtain the lowest χ scores between the experimental, ΔHexp(i), and theoretical, Q(i) − Q(i − 1), values using a homemade procedure coupled with the Marquardt-Levenberg algorithm (26) implemented in the Gnuplot program (available on the World Wide Web).
where Q(i) is the estimated heat release after i succeeding injections, and S100[ijk](i) are populations of variously Ca2+-loaded S100A1 forms after the ith injection, calculated according to the equations above. The values of stability constants (k100, k110, and k11n) and accompanying heats of binding (H100, H110, and H11n) were optimized to obtain the lowest χ scores between the experimental, ΔHexp(i), and theoretical, Q(i) − Q(i − 1), values using a homemade procedure coupled with the Marquardt-Levenberg algorithm (26) implemented in the Gnuplot program (available on the World Wide Web).
Recombinant Human Apo-S100A1 and Apo-S100A1-NO for Structural Studies in Solution
13C,15N-double-labeled recombinant human S100A1 was obtained as described previously (27). Briefly, the synthetic gene was cloned into pET-30a+ plasmid and expressed in E. coli utilizing a T7 expression system. The bacteria were grown in M9 medium containing (15NH4)2SO4 as the sole nitrogen source and 13C-labeled glucose as the sole carbon source. Classical purification methods with no affinity tags were used for protein purification to avoid any changes to the protein sequence other than S-nitrosylation. Expression products were isolated using precipitation by ammonium sulfate and purified by reversed-phase HPLC on a semipreparative Vydac C18 column. Denaturing HPLC, performed under acidic pH, was used to obtain the highest homogeneity of S100A1 isoforms (27). Two forms of S100A1, one with the sequence strictly corresponding to its gene sequence and another one with an additional initiator methionine at the N terminus, were obtained as partly overlapping HPLC peaks and were used for NMR experiments. Correct masses were confirmed by ESI-MS using a Q-TOF Premier spectrometer (Waters Inc.). Purified proteins were successfully refolded prior to NMR experiments. Both 13C-,15N-labeled apo-S100A1 and apo-S100A1-NO variants have almost identical CD spectra characteristic of highly helical S100 proteins. The homodimeric state of apo-S100A1 and apo-S100A1-NO was confirmed by a gel filtration method (data not shown).
NMR Spectroscopy and Resonance Assignments
NMR samples of 13C,15N-double-labeled human apo-S100A1 and apo-S100A1-NO proteins were prepared by dissolving 0.8 mm protein in 25 mm Tris-d11 buffer containing 150 mm NaCl (pH 7.2, uncorrected value). All spectra were acquired at 298 K on a Varian VNMRS 800 NMR spectrometer (Varian Inc., Palo Alto, CA) equipped with four channels, z axis Performa II gradient unit, and triple 1H/13C/15N cryogenic probehead with inverse detection. The sensitivity-enhanced detection procedures (28) were used jointly with States-TPPI quadrature detection (29). Chemical shifts were referenced with respect to external sodium 2,2-dimethyl-2-silapentane-5-sulfonate and processed with NMRPipe software (30). The 13C and 15N resonances were indirectly referenced using the 0.251449530 and 0.101329118 ratios for 13C and 15N, respectively (31). All spectra were analyzed with Sparky (32) and CARA (see the Swiss NMR website) programs.
Assignments of 1H, 13C, and 15N backbone resonances in a sequence-specific manner were obtained by standard methods based on the analysis of three-dimensional heteronuclear HNCACB, CBCA(CO)NH, HNCA, and HN(CO)CA spectra (33). Obtained chemical shifts were additionally confirmed by inspection of the three-dimensional 15N-edited NOESY-HSQC data set. 1H and 13C resonance signals in aliphatic side chains were assigned using three-dimensional HBHA(CO)NH, (H)CCH-TOCSY, and 13C-edited NOESY-HSQC experiments. 1H and 13C resonances in aromatic side chains were assigned based on the analysis of three-dimensional 13C-edited NOESY-HSQC spectrum recorded with parameters tuned to aromatic carbons.
Experimental Restraints and Three-dimensional Structure Calculations
The structure calculations for both variants of human apo-S100A1 protein were performed with CYANA software (version 3.0), which contains a specific module for automatic assignment and calibration of resonance cross-peaks in multidimensional three-dimensional/four-dimensional NOESY spectra suitable for protein homodimers. The assignment procedure yielded 2964 and 2916 distance constraints for reduced and S-nitrosylated forms, respectively, which were used for three-dimensional structure CYANA program suite evaluation. In this stage of calculations, 142 and 148 stereo-specific assignments were defined for apo-S100A1 and apo-S100A1-NO by the program GLOMSA included in the CYANA work package (34). Afterward, automatically assigned distance constraints were manually checked to eliminate some incorrect or ambiguous NOEs. Assigned NOE constraints were supplemented by 328 or 312 backbone torsion angle restraints defined from the analysis of backbone chemical shifts with TALOS+ software (35). Finally, 168 or 124 1DNH RDCs extracted from the 1H-15N in-phase/anti-phase experiment (36) performed in two different media were added as long range distance constraints for apo-S100A1 and apo-S100A1-NO, respectively. The axial (Aa) and rhombicity (Ar) components of alignment tensors were defined from histograms of the 1DHN dipolar couplings. As a result, 20 of 200 calculated structures characterized by lowest target function were selected for additional refinement in explicit solvent. The refinement procedure was conducted in water solution with the Yasara software package (37) using the Yasara2 force field. Parameterization of S-nitrosocysteine used for structure calculations of apo-S100A1-NO protein was taken directly from the force field. Table 1 presents all of the experimental data used for solving the high resolution three-dimensional structure of both variants of human apo-S100A1 protein. The quality analyses of ensembles were performed with WhatIf (38) and Procheck (39) software. Final structures were deposited in the PDB data bank under Protein Data Bank code 2LLU and 2LLT for apo-S100A1 and apo-S100A1-NO, respectively. Figures presenting the three-dimensional structures of proteins were generated with the Chimera program (40).
Structural constraints used for solution high resolution three-dimensional structures and statistical analysis of ensembles of apo-S100A1 and apo-S100A1-NO proteins
RESULTS
S100A1 Protein Is Endogenously S-Nitrosylated in PC12 Pheochromocytoma Cells
The PC12 pheochromocytoma cells have been widely used as a model system to study molecular mechanisms of S100A1 protein activity. They are known to express S100A1 at a high level (23). PC12 cells not stimulated with nitric oxide donors or other compounds were used to determine whether endogenous S-nitrosylation of S100A1 protein might be observed in a cellular system. The whole cell lysate of confluent, unstimulated PC12 cells was treated with the biotin switch method, which is an established procedure to study protein S-nitrosylation (41). Using BSM, the S-nitrosylated protein cysteines are selectively converted to S-biotinylated cysteines. Thus, BSM combined with neutravidin affinity chromatography allows one to selectively fish out the proteins previously endogenously S-nitrosylated inside PC12 cells. A control experiment in which the ascorbate reduction of S–NO bonds is not included in BSM has been used to obtain proteins that bind nonspecifically to neutravidin resin. All obtained protein fractions were analyzed by Western blot using a specific anti-S100A1 antibody (Fig. 1). An identical procedure was used for solutions of recombinant S100B protein selectively S-nitrosylated on the C-terminal Cys84 and not on Cys68, which was present as a free thiol. The control S100B-NO protein was detected using Western blot analysis only in the fractions expected for BSM (supplemental Fig. S2). S100A1 was detected only in the PC12 protein fractions enriched using complete BSM (Fig. 1, lane 9) and not in the fractions obtained using BSM without ascorbate reduction of the S–NO bond (Fig. 1, lane 8). This confirms that S-nitrosylation is an endogenous post-translational modification of S100A1 protein present in PC12 cells.
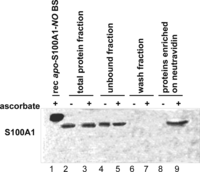
Detection of endogenous S-nitrosylation of apo-S100A1 protein in PC12 pheochromocytoma cell line using the biotin switch method combined with anti-S100A1 Western blot. Lane 1, chemically S-nitrosylated, recombinant human apo-S100A1-NO protein eluted from neutravidin resin after BSM enrichment (rec apo-S100A1-NO BS); lane 2, total protein fraction (control BSM without Asc) before affinity enrichment on neutravidin; lane 3, total protein fraction (full BSM) before affinity enrichment on neutravidin; lane 4, protein fraction unbound to neutravidin (control BSM without Asc); lane 5, protein fraction unbound to neutravidin (full BSM); lane 6, wash fraction (control BSM without Asc); lane 7, wash fraction (full BSM); lane 8, proteins enriched on neutravidin resins (control BSM without Asc); lane 9, proteins enriched on neutravidin resins (full BSM).
Influence of S-Nitrosylation on Ca2+ Binding to S100A1
Ca2+ binding is a known prerequisite for the interaction of S100A1 with a variety of physiologically important targets (42, 43). Notwithstanding, the calcium affinities measured for S100A1 at higher ionic strength are too low to allow the formation of an S100A1-Ca2+ complex during calcium influx in vivo (17, 44). We were interested in whether the post-translational S-nitrosylation detected by us in PC12 cells could be a factor that modifies calcium binding to S100A1 protein. To address this question, we used ITC to study Ca2+ binding to recombinant human S100A1 and its S-nitrosylated derivative. Ca2+ titration experiments were performed for S100A1 and S100A1-NO protein solutions (40 μm) in TES buffer, pH 7.4, containing 150 mm NaCl. Fig. 2 presents the comparison of ITC data collected for both forms of S100A1 protein. The figure demonstrates that for S100A1-NO, the Ca2+-loaded complex is fully formed after a much smaller number of CaCl2 injections. Thus, S-nitrosylation significantly increases the affinity of S100A1 toward Ca2+ ions. Binding of Ca2+ ion is an endothermic process for all studied S100A1 samples. For S100A1-NO, the binding of Ca2+ is stronger than for S100A1 but is accompanied by a lower heat flow. This leads to a conclusion that S-nitrosylation induces a conformational transition of S100A1 that, in terms of conformational entropy, significantly favors a Ca2+-bound state of the protein.
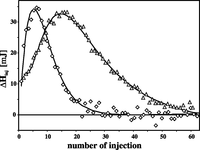
Comparison of calorimetric profiles recorded for S100A1 and S100A1-NO. Shown are ITC titrations of S100A1 (triangles) and S100A1-NO (diamonds) protein solutions (40 μm) in TES buffer, pH 7.4, containing 150 mm NaCl. Solid lines represent fitted models (see Table 2 for thermodynamic parameters of Ca2+ binding).
Ca2+ binding to S100A1 protein has been analyzed previously using flow dialysis (17, 19). A model of four Ca2+ ions sequentially binding to a dimer of S100A1 was proposed by the authors to provide the best fit to experimentally obtained
data. The previously published model and several other theoretical models based on the binding of two Ca2+ ions to a protein monomer were used to calculate the thermodynamic parameters of Ca2+ binding to S100A1 under our experimental conditions, Unfortunately, our data were not properly reconstructed by any such
models. Thus, we used a method originally proposed by Job (45) to roughly estimate the stoichiometry of the formed S100A1-Ca2+ complexes. Job plots (supplemental Fig. S3) represent the relation of values that are calculated based on the two formulas described in Equation 8, with Qtot being the cumulative heat flow measured upon titration of S100A1 with Ca2+ ions and [CaCl2] and [S100A1] being the concentrations of reactants.
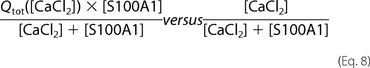 For the control experiment, in which Ca2+ was titrated into an S100A1 solution without NaCl, the maximum of the Job plot is located at ∼0.6. This is equivalent to
two Ca2+ binding sites per S100A1 monomer and is in agreement with the number of EF-hand domains in S100A1. In our experiments at
higher ionic strength, the maxima of Job plots are unequivocally shifted toward higher stoichiometries for both S100A1 and
S100A1-NO. The plots suggest that at least four Ca2+ ions are bound per protein monomer. Thus, to derive Ca2+ binding constants for S100A1 and S100A1-NO in physiological ionic strength buffers, we extended the sequential model of binding
of two Ca2+ cations to S100 proteins by a third type of binding site of an unknown occupancy n per monomer. The best fit between models and experimental data for both forms of S100A1 was obtained for n = 2, indicating that S100A1 in 150 mm NaCl solution apparently binds four Ca2+ cations per monomer. Our calculations showed substantial differences in the determined thermodynamic parameters and order
of Ca2+ binding between S100A1 and S100A1-NO (Table 2). A much higher cooperativity in binding of the first two Ca2+ ions is observed for S100A1-NO than for S100A1. Introduction of the S-NO group leads to simultaneous binding of two Ca2+ cations to S100A1-NO with a relatively high apparent stability constant of 3.1 × 107 m−2. Under the experimental conditions in this work, S100A1 binds the first two Ca2+ ions with an effective affinity approximately 1 order of magnitude lower (∼4.5 106 m−2). Moreover, the total entropy gain upon binding of the first two Ca2+ ions to S100A1-NO is at least 2-fold larger than that observed for S100A1, 49.6 versus 20.2 cal/mol/K, respectively. The change of entropy upon Ca2+ binding to the second site is unfavorable in S100A1 (−5 cal/mol/K), whereas for S100A1-NO, the average entropy binding for
one of the two Ca2+ ions (24.8 cal/mol/K) closely corresponds to the value characterizing the first binding site in S100A1 (25.4 cal/mol/K).
Such differences in the values of entropic terms suggest that S-nitrosylation induces some structural reorganization of S100A1, resulting in a substantial increase of affinity of the second
Ca2+ ion.
For the control experiment, in which Ca2+ was titrated into an S100A1 solution without NaCl, the maximum of the Job plot is located at ∼0.6. This is equivalent to
two Ca2+ binding sites per S100A1 monomer and is in agreement with the number of EF-hand domains in S100A1. In our experiments at
higher ionic strength, the maxima of Job plots are unequivocally shifted toward higher stoichiometries for both S100A1 and
S100A1-NO. The plots suggest that at least four Ca2+ ions are bound per protein monomer. Thus, to derive Ca2+ binding constants for S100A1 and S100A1-NO in physiological ionic strength buffers, we extended the sequential model of binding
of two Ca2+ cations to S100 proteins by a third type of binding site of an unknown occupancy n per monomer. The best fit between models and experimental data for both forms of S100A1 was obtained for n = 2, indicating that S100A1 in 150 mm NaCl solution apparently binds four Ca2+ cations per monomer. Our calculations showed substantial differences in the determined thermodynamic parameters and order
of Ca2+ binding between S100A1 and S100A1-NO (Table 2). A much higher cooperativity in binding of the first two Ca2+ ions is observed for S100A1-NO than for S100A1. Introduction of the S-NO group leads to simultaneous binding of two Ca2+ cations to S100A1-NO with a relatively high apparent stability constant of 3.1 × 107 m−2. Under the experimental conditions in this work, S100A1 binds the first two Ca2+ ions with an effective affinity approximately 1 order of magnitude lower (∼4.5 106 m−2). Moreover, the total entropy gain upon binding of the first two Ca2+ ions to S100A1-NO is at least 2-fold larger than that observed for S100A1, 49.6 versus 20.2 cal/mol/K, respectively. The change of entropy upon Ca2+ binding to the second site is unfavorable in S100A1 (−5 cal/mol/K), whereas for S100A1-NO, the average entropy binding for
one of the two Ca2+ ions (24.8 cal/mol/K) closely corresponds to the value characterizing the first binding site in S100A1 (25.4 cal/mol/K).
Such differences in the values of entropic terms suggest that S-nitrosylation induces some structural reorganization of S100A1, resulting in a substantial increase of affinity of the second
Ca2+ ion.
Thermodynamic parameters for calcium binding estimated from isothermal titration of protein solution with CaCl2
For both forms of S100A1, in accordance with stoichiometry derived from Job plots, sequential binding of Ca2+ by two binding sites is followed by binding to another two cations by yet unidentified site(s).
The additional two Ca2+ binding sites in S100A1, detected by us uniquely in higher ionic strength buffers, bind Ca2+ ions stronger than analogous sites in S100A1-NO. However, moderate values of stability constants, falling in the order of 103 m−1, suggest that those sites are probably functional for neither of the S100A1 forms (Table 2 and supplemental Fig. S3). The lack of involvement of the Cys85 side chain in direct Ca2+ binding by EF-hand loops makes it difficult to understand how the addition of an S-nitroso group could alter the Ca2+ binding properties of S100A1. Therefore, to better understand this phenomenon, we performed detailed, differential structural analyses of apo-S100A1 and apo-S100A1-NO in solution.
Sequence-specific Resonance Assignment and Three-dimensional Structure Calculations
Three-dimensional NMR experiments were performed under identical experimental conditions for apo-S100A1 and apo-S100A1-NO. For both protein forms, a standard approach based on the analysis of three-dimensional HNCA/HN(CO)CA and HNCAB/CBCA(CO)NH spectra was used to assign 1H, 13C, and 15N backbone resonances in a sequence-specific manner. The chemical shift of 13Cβ nuclei of Cys85 in apo-S100A1 was 32.4 ppm, a typical value for the reduced state of a thiol group in cysteine (46). In apo-S100A1-NO, the chemical shifts for side-chain nuclei of Cys85 could not be determined due to signal suppression, most probably caused by the proximal π electron system of the S-NO moiety. This effect is also reflected in increased line widths observed for resonances from the neighboring residues Asn86 and Asn87 (supplemental Fig. S4). The side chains 1H and 13C resonances were assigned on the basis of three-dimensional HCCH-TOCSY and 13C-edited NOESY spectra. Finally, more than 96% of all chemical shifts were defined and deposited in the Biological Magnetic Resonance Bank (BMRB accession numbers 18089 and 18088 for apo-S100A1 and apo-S100A1-NO protein, respectively).
The position of α-helices within individual subunits has been initially deduced from chemical shift analysis of backbone resonances with TALOS+ software (35). Later, obtained results were confirmed by the existence of characteristic NOESY patterns on 15N-edited NOESY-HSQC spectra (supplemental Fig. S5A). For apo-S100A1 protein, four α-helices were defined: Glu3–Ala17, Lys30–Glu40, Val51–Glu63, and Phe71–Ala86. Similarly to other S100 protein structures in the apo state, a short, one-turn α-helix was observed for residues Phe44–Ala47 in the “hinge” region that connects helices II and III (47). A short anti-parallel β-strand motif responsible for interactions between the N- and C-terminal EF-hand Ca2+-binding loops was identified among Leu28–Ser29 and Glu68–Val69. Similar secondary structure was defined for apo-S100A1-NO with the exception of helix IV, which is one turn shorter (Phe71–Ala84), and the α-helical pattern in the hinge region elongated by two residues (Ser42–Asp46) (supplemental Fig. S5B).
The CYANA program suite, together with a module enabling automatic assignments of NOE distance constraints for protein homodimers, was used for initial elucidation of high resolution three-dimensional structures of apo-S100A1 and apo-S100A1-NO. Calculations yielded 2964 distance constraints for apo-S100A1 (676 intraresidual, 874 sequential, 818 medium range, 394 long range, and 202 intersubunit) and 2916 distance constraints for apo-S100A1-NO (728 intraresidual, 774 sequential, 774 medium range, 420 long range, and 220 intersubunit) (Table 1). The angular restraints (328 and 312) for backbone ϕ and ψ torsion angles were derived by TALOS+, yielding a good prediction of backbone conformation for 82 and 78 residues in apo-S100A1 and apo-S100A1-NO, respectively, and were included in the further calculation procedure as additional restraints.
RDC data provide important long range distance constraints that considerably increase the accuracy of evaluated three-dimensional structures. Due to symmetry requirements in a homodimeric protein, the alignment of one of the non-degenerative principal axes becomes collinear with the symmetry axes of the molecule (48). To solve this degeneracy, the RDC data sets for homodimers should be acquired in at least two different media (49). Thus, we extracted the experimental RDC long range distance constraints from two-dimensional 1H-15N in-phase/anti-phase NMR experiments (36) conducted in two kinds of oriented media, Pf1 phages (50) and dimyristoyl phosphatidylcholine/dihexanoyl phosphatidylcholine bicelles (51). Analysis of acquired data yielded 184 and 142 1DHN RDCs for apo-S100A1 and apo-S100A1-NO, respectively, which were included in the three-dimensional structure evaluation procedures. As a result, the quality factors equal to 0.23 in Pf1 phages and 0.46 in bicelle media for the apo-S100A1 and to 0.26 in Pf1 media and 0.30 in bicelle media for the apo-S100A1-NO have been reached, proving that calculated three-dimensional structures are in agreement with the experimental long range distance constraints.
Comparative Analysis of NMR Data for Human Apo-S100A1 and Apo-S100A1-NO
Initial analysis of chemical shift perturbations (Δδtot), calculated as a weighted combination of 1HN, 15N, 13C′, and 13Cα chemical shifts (52), revealed that S-nitrosylation of Cys85 in S100A1 protein mostly affects the linker residues (Leu41–Val51) and the C-terminal part of helix IV (Leu81–Asn87) (Fig. 3). However, Δδtot values in those regions lie in the range of 0.1–0.35 ppm and are smaller than those previously observed between apo-S100A1 and apo-S100A1-βME forms (53). Alterations of backbone conformation upon S-nitrosylation were further confirmed by analysis of backbone mobility with the random coil index approach (54). Both variants of apo-S100A1 protein demonstrate small but notable variations in random coil index and S2 in the same regions of the apo-S100A1 three-dimensional structure (supplemental Fig. S6).
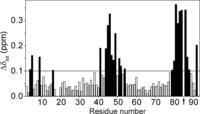
Chemical shift perturbations as a consequence of Cys85 S-nitrosylation. The combined chemical shifts differences (Δδtot) between apo-S100A1 and apo-S100A1-NO variants were calculated as Δδtot = ((ΔδHN)2 + (0.154·ΔδN)2 + (0.341·ΔδCO)2 + (0.276·ΔδCα)2)½ (52). The position of S-nitrosylation is marked with an arrow. Black bars correspond to residues with Δδtot values higher than 0.1 ppm.
S-Nitrosylation Induces Structural Changes in Apo-S100A1 Protein
The S100 homodimers form a tight X-type intersubunit interface stabilized by hydrophobic interactions formed by residues from helix I and IV from both monomers (supplemental Fig. S9) (55). Although the global fold in both variants of apo-S100A1 is the same, detailed analysis demonstrated structural differences that are not limited to the area close to the S-nitrosylated Cys85 (Fig. 4, A–C). One of the most interesting effects of S-nitrosylation of S100A1 is the restructuring of the linker region between two EF-hand motifs. Introduction of an S-NO group to the thiol group of Cys85 results in an extension of the α-helix length from Phe44–Ala47 to Ser42–Asp46 (supplemental Fig. S5), making the linker connecting helices II and III much more rigid. Such structural alterations are supported by differences observed in NOE contacts between residues located in the linker and the C-terminal part of helix IV presented on a two-dimensional map of experimentally observed NOE contacts between side-chain protons in both apo-S100A1 forms (supplemental Fig. S8). For example, contacts between residues Asp50 and Trp90, Ala53 and Trp90, Lys49 and Asn86, and Lys49 and Cys85 are indicative for apo-S100A1, whereas only two interactions were noted in this region for apo-S100A1-NO: Asp50-Cys85 and Asp50-Ala84 (Fig. 4A). The two most varied regions on the NOE contact map (supplemental Fig. S8, regions A and B) reflect the biggest structural differences at two sites of S100A1 dimer, which form the interface for interactions with target proteins and small compounds (42, 56).
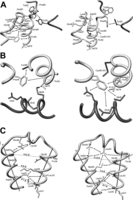
Fragments of high resolution three-dimensional structures of reduced (left) and S-nitrosylated (right) variants of human S100A1 protein. A, the part of the structure showing the C terminus, hinge region, and N-terminal Ca2+-binding loop. B, differences in the contacts of Phe44 aromatic side chain with the C-terminal part of helix IV and central part of helix I′. C, maps of contacts observed between residues from helix II, helix III, and the hinge region.
Introduction of a bulky S-NO group to the side chain of Cys85 forces a rearrangement of side chains of C-terminal aromatic residues (Phe88, Phe89, and Trp90) in the direction away from the linker region but toward the EF-hand loops of the adjacent subunit. Nevertheless, in both versions of apo-S100A1, a local hydrophobic cluster is formed, which includes side chains of aromatic residues Phe88, Phe89, and Trp90 from one monomer together with the phenyl group of Tyr26 located in the N-terminal Ca2+ binding loop of the other subunit. Similar intersubunit NOE contacts that indicate an interaction between these protein sites have been detected previously in apo-S100A1 and its mixed disulfide forms (18, 53). Another aromatic residue, Phe44, is the central residue of the linker region. Inspection of the aromatic two-dimensional 1H-13C HSQC spectrum showed that the most pronounced changes in chemical shifts of aromatic protons are detected in the side chain of Phe44 (Fig. 5A). Processed three-dimensional 13C-edited NOESY-HSQC data sets exhibited several NOE contacts between protons of Phe44 (1Hϵ and 1Hz) and helix I′ residue Glu5 (1Hα), which are characteristic exclusively of the apo-S100A1-NO form (Fig. 5B). Comparison of the distance constraints used for high resolution three-dimensional structure calculations revealed the existence of eight long range contacts between protons from Phe44 and Glu5, Met8, and Glu9 from helix I′ of the adjacent subunit for the S-NO variant of the human S100A1 protein (Fig. 4B and supplemental Fig. S8, region B). Thus, as a consequence of S-nitrosylation, the aromatic side chain of Phe44 is rotated in the direction of helix I′ and the side chain of Cys85. This leads to an extension of a short α-helix in the hinge region of S100A1 (Fig. 4B).
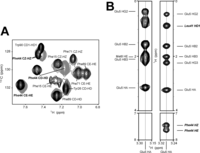
NMR data on aromatic side chains in reduced and S-nitrosylated variants of apo-S100A1 protein. A, overlay of two-dimensional aromatic 1H-13C HSQC spectra acquired for apo-S100A1 protein in reduced (black) and S-nitrosylated (gray) form. B, two-dimensional 1H-1H planes of three-dimensional 13C-edited NOESY-HSQC spectra taken at the frequency corresponding to 1Hα Glu5. 1Hα Glu5-1Hϵ Phe44 and 1Hα Glu5-1Hz Phe44 are clearly observed in the case of apo-S100A1-NO.
Interhelical angles for all pairs of helices were calculated for the structures of human apo-S100A1 and apo-S100A1-NO solved in this work (Table 3). S-Nitrosylation does not perturb the intersubunit interface formed by helices I–I′ and IV–IV′. At the same time, we observe a significant change from 123° up to 140° in the angle between antiparallel helices II and III in apo-S100A1-NO. A similar effect was discussed previously for a monomeric S100 protein, calbindin D9K, where after a Phe36 → Gly mutation in the linker region, the interhelical angles change from 112.5 to 170.4°, positioning the helices II and III in a similar way as those in calmodulin (57). The different orientation of helices II and III is reflected in the divergent NOE contacts observed in that region (Fig. 4C and supplemental Fig. S8). Only a few NOEs between residues from helices II and III (Lys30, Lys31, Leu33, and Lys34–Leu61) were observed in apo-S100A1. The contacts between Gln38 (helix II) and Met58 and Asp62 (helix III) are unique and detected only in apo-S100A1-NO.
Interhelical angles observed in three-dimensional structures of S100A1 and S100B proteins calculated with supporting RDC long range distance constraints
S100 family proteins are usually stable homodimers with high dimerization constants (58). The dimeric interface constitutes of a large hydrophobic surface. Our experimental data exhibited more than 200 intersubunit contacts, 202 and 244 NOEs for apo-S100A1 and apo-S100A1-NO, respectively (Table 1). Both apo-S100A1 and apo-S100A1-NO are very stable in solution. The proteins are not prone to degradation phenomena even for several months of solution storage. The NMR samples were stored in a fully deuterated buffer for more than 6 months at 277 K. Nevertheless, two-dimensional 1H-15N HSQC spectra acquired after such a long period clearly showed preserved amide proton signals, which were assigned to residues from helix I (Met8, Glu9, Leu11, Ile12, and Phe15) and helix IV (Tyr74, Val75, Val76, Leu77, Val78, Ala79, Ala80, Leu81, and Thr82) and hydrophobic residues in helix II (Leu33, Leu36, and Leu37) (Fig. 6A). Retained signals from amide proton for residues Ile12, Leu33, Leu36, Leu37, Val76, Leu77, Val78, and Leu81 had higher intensities in the spectrum of apo-S100A1-NO, suggesting a stronger protection from the solvent and a more powerful interaction between subunits for apo-S100A1-NO in comparison with apo-S100A1. Arrangement of these residues within the dimeric apo-S100A1 structure exhibited a unique inner hydrophobic core with two clearly distinguished regions (supplemental Fig. S7). First are residues that do not show any visible changes upon S-nitrosylation in the location of amide resonances in 1H-15N HSQC spectra (Leu33, Leu36, and Val75–Ala79). These residues do not undergo backbone rearrangement and constitute a common 1-connected hydrophobic core and contain the framework of helices II, IV, IV′, and II′ (Fig. 6B, shown in yellow). Organization of this core was found almost identical in both protein forms. On the other hand, about 15 other residues, with the amide proton signal retained from exchange, have significantly altered chemical shifts in the spectrum of apo-S100A1-NO, indicating a change in their conformation (Fig. 6B, shown in orange). This modulated part of the hydrophobic core is located in the close vicinity of the aromatic linker residue Phe44 (supplemental Fig. S10). Most probably, the latter residues serve to transmit the S-NO signal to other peripheral residues and sites of the S100A1 protein.

Hydrophobic core packing in apo-S100A1 and apo-S100A1-NO proteins. A, overlay of two-dimensional 1H-15N HSQC NMR spectra acquired at 298 K on a Varian VNMRS 800 NMR spectrometer for apo-S100A1 (gray) and apo-S100A1-NO (black) after 6 months of storage at 4 °C in D2O solution. B, ribbon representation of the structures of apo-S100A1 (top) and apo-S100A1-NO (bottom). Side chains of residues Phe44, Cys85, and Cys85-NO are presented as sticks, and van der Waals radii for residues with highly protected amide protons (A) with negligible or large chemical shift perturbations are shown in yellow and orange, respectively.
DISCUSSION
Analyses of evolutionary changes in sequences of proteins have recently led to a hypothesis that some protein cysteine thiols have evolved in proteins to serve as sensors of endogenous oxidizing molecules, in particular nitric oxide (59). It is now widely acknowledged that nitric oxide may regulate protein activity through selective formation of covalently S-nitrosylated cysteines (60). Nine members of the S100 protein family have a conserved cysteine residue at the C terminus. All of these proteins belong to a closely phylogenetically related subgroup with 13 absolutely conserved amino acid positions (supplemental Fig. S1). Our study illustrates that an important member of this subgroup, the S100A1 protein, is endogenously S-nitrosylated at the C-terminal Cys85.
The three-dimensional structure of apo-S100A1-NO presented in this work is one of only a few structures solved to date for S-nitrosylated proteins. Due to the lability of the S-nitroso group under x-ray conditions, only four structures of S-NO proteins have been solved by x-ray crystallography (61,–,64). High resolution three-dimensional structure solved by NMR spectroscopy has been published only for recombinant S-nitrosylated Ras protein (65). Although S-NO formation regulates the biological activity of Ras protein, it did not significantly alter the its three-dimensional structure in solution.
Data presented in this work showed that apo-S100A1 protein in a Tris buffer containing 150 mm NaCl appears as a very stable noncovalent homodimer of four-helix bundles. This is similar to S100 protein structures solved under other experimental conditions (47, 66). The overall structure of S100A1 is not grossly modified by S-nitrosylation. The integrity and stability of the apo-S100A1-NO homodimer remains similar. Side-by-side analysis of NMR data suggests that S-nitrosylation of Cys85 reshapes structural elements in S100A1 at sites distant from the modified residue. This is in agreement with the previously published hypothesis that S100 protein dimers should be considered as single globally cooperative structural units in which structural perturbations may be found far from the site of modification (57). S-Nitrosylation influences the conformation of the hydrophobic core and changes the packing and relative orientation of helices II and III. The two regions with the biggest S-NO-induced conformational rearrangements are the linker region and the two helices III and IV from the C-terminal EF-hand of S100A1. These regions are exposed to solvent after transition of S100A1 from the “closed” to the “open” conformation after calcium binding (66, 67). In detail, S-nitrosylation forces exposure to solvent of most C-terminal residues Cys85–Ser93. This protein fragment has been shown to be particularly important for Ca2+-dependent target binding of S100A1 protein with, for example, TRTK-12 peptide and fragments of GFAP and p53 (68, 69). Residues Phe88–Ser93 in S100A1 were also identified as the Ca2+-dependent binding site for the antiallergic drug Amplexanox (56). Thus, S-nitrosylation may directly influence target recognition by S100A1 in vivo.
Calcium affinity of unmodified S100A1 is too low to form the calcium-bound protein necessary for interaction with various important target molecules (44). S-Nitrosylation of S100A1 increases the affinity and cooperativity of binding of the first two Ca2+ cations (Table 2). S-NO-dependent cell signaling may directly influence the population of calcium-bound S100A1 at physiological calcium levels and enhance the physiologically important calcium-dependent interactions. Nevertheless, in our structural studies, no experimental evidence has been observed for S-NO-induced conformational variations in the backbone or side chains of residues responsible for Ca2+ ion coordination. Additionally, ITC data suggest that entropic effects favor the calcium-bound form of S100A1-NO. This is consistent with the observed S-NO-induced exposure of aromatic residues, which must lead to different hydration of the protein. The loss of side-chain entropy due to formation of a more rigid linker loop most probably leads to enhanced flexibility of other protein regions, as previously observed during calcium binding to calbindin D9k (70). It is worth noting that S-NO-induced elongation of the helical element in the hinge region shown in this study for homodimeric apo-S100A1 resembles that described for the monomeric calbindin D9k mutant P43MG (71). In both cases, a short helix in the weakly structured linker is elongated and anchored to the hydrophobic core and correlates with increased Ca2+ affinities of the modified proteins (71). Another study performed on dimeric S100B protein has revealed that Ca2+ binding increases upon binding of a peptide fragment of its target protein, p53 (72). Structural NMR analysis has demonstrated that binding of p53 peptide influences conformation of the hinge region in S100B. The above examples suggest that structural perturbations of the linker connecting two EF-hand domains in S100 proteins, through different molecular mechanisms, may lead to variations in their calcium binding affinity. In this work, we suggest yet another molecular mechanism of modification of the linker region in an S100 protein, which is based on the reactivity of a conserved cysteine residue. Cysteine thiols that do not form disulfide bonds often contribute to protein structure stability by forming conventional hydrogen bonds. If the cysteine is in a hydrophobic environment, it is often engaged in electrostatic interactions with aromatic residues (73, 74). The covalent addition of nitric oxide to the thiol group changes the geometry of the thiol-aromatic interaction. It has been proposed previously that reshaping of the aromatic-thiol interactions may be a generic mechanism by which S-nitrosylation alters protein structure and function (59). Based on comparison of high resolution structures solved in this work, we suggest that the molecular mechanism leading to overall structural changes in apo-S100A1-NO and modulation of Ca2+ affinity is the difference in the mutual arrangement of residues Cys85 and Phe44 from adjacent S100A1 monomers. The Cys85/Phe44 rearrangement directly fine tunes the conformation of the hydrophobic core of S100A1, which widely spans the whole protein dimer and transmits the S-NO signal to peripheral residues of the protein (Fig. 6B).
Recently, a hypothesis that the less structured regions of S100 proteins have many features of intrinsically disordered proteins (IDPs) has been published (75). The IDPs lack any rigid three-dimensional structure under physiological conditions, rather existing as dynamic ensembles of interconverting structures. The disordered regions in IDPs are responsible for the functional diversity of the whole proteins that complement the functions of the ordered protein regions. The activity of IDPs is often regulated by post-translational modifications of residues directly in the unstructured regions. In our opinion, S-nitrosylation of S100A1 may be considered as a mechanism of regulation of an IDP region that is realized not by direct modification of the disordered fragment but by an indirect through-space interaction of the cysteine side chain with the aromatic ring of Phe44.
Alignment of S100 proteins sequences (supplemental Fig. S1) clearly shows that an aromatic residue is strictly conserved in proteins that have a C-terminal cysteine in their primary sequence. This supports our idea of the importance of the NO-sensitive, thiol-aromatic conformational switch, which may be a more universal molecular mechanism regulating the biological activity of this subfamily of S100 proteins. The functional consequences of endogenous S-nitrosylation of S100A1 have yet to be established. S100A1 gene therapy is in clinical trials to rescue heart failure (11). S-nitrosylation plays an important role in NO/redox-based signaling in physiologic regulation of cardiac contractility as well as in pathophysiology of heart failure (9, 76). Nitric oxide can, through S-nitrosylation, modulate the L-type Ca2+ channel, ryanodine receptor (RyR), and sarcoplasmic reticulum ATPase (SERCA2a), which all participate in Ca2+ handling and contractile performance in cardiomyocytes. S100A1 interacts with both RyR and SERCA2a, which is reflected in improved Ca2+ cycling and diminished Ca2+ leakage during diastole (10). At increased levels of calcium entry, S-nitrosylated protein would more quickly adopt a holo conformation and thus more strongly interact with RyR and SERCA, which would be reflected through better cardiomyocyte performance. On the other hand, S100A1 could transfer the NO moiety to other proteins that are known to be regulated by S-nitrosylation (RyR, SERCA2a, etc.) and hence influence cardiac EC coupling and other cellular processes (76). It is most probable that S-nitrosylation does not act independently to regulate the function of S100A1 but rather acts in complex associations with other factors (e.g. binding of the target protein). Although our knowledge of the effect of S-nitrosylation on the functioning of human S100A1 is far from complete, the data presented in this work should be considered in the process of development of S100A1-based therapies.
Footnotes
-
↵* This work was supported by Polish Ministry of Education Grant 3P04A 01424 and Slovenian Research Agency and Ministry of Higher Education, Science, and Technology of Republic of Slovenia Program P1-0242 (to M. L. Ž.). This work was also supported in part by the EN-FIST Center of Excellence (to I. Z.), East-NMR FP7 Project (Contract 228461), and Transnational Access Program and Innovative Economy Program Grant POIG.01.01.02-00-048/09 co-financed by European Union resources (to A. W.).
-
↵
 This article contains supplemental Figs. S1–S10.
This article contains supplemental Figs. S1–S10.
-
The atomic coordinates and structure factors (codes 2LLT and 2LLU) have been deposited in the Protein Data Bank (http://wwpdb.org/).
-
↵4 The abbreviations used are:
- apo-S100A1
- human S100A1 protein in apo-form (without Ca2+ ions)
- apo-S100A1-NO
- human S100A1 protein in apo-form with S-nitrosylated Cys85
- GSNO
- S-nitrosoglutathione
- ITC
- isothermal titration calorimetry
- BSM
- biotin switch method
- Asc
- ascorbate
- HSQC
- heteronuclear single quantum correlation spectroscopy
- biotin-HPDP
- N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide
- TES
- 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid
- RDC
- residual dipolar coupling
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- IDP
- intrinsically disordered protein
- RyR
- ryanodine receptor.
- Received September 10, 2012.
- © 2012 by The American Society for Biochemistry and Molecular Biology, Inc.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵










