
Three-layered control of mRNA poly(A) tail synthesis in Saccharomyces cerevisiae
- Matti Turtola1,
- M. Cemre Manav2,
- Ananthanarayanan Kumar2,6,
- Agnieszka Tudek3,
- Seweryn Mroczek4,5,
- Paweł S. Krawczyk5,
- Andrzej Dziembowski4,5,
- Manfred Schmid1,
- Lori A. Passmore2,
- Ana Casañal2,7 and
- Torben Heick Jensen1
- 1Department of Molecular Biology and Genetics, Aarhus University, 8000 Aarhus C, Denmark;
- 2Medical Research Council Laboratory of Molecular Biology, Cambridge CB2 0QH, United Kingdom;
- 3Institute of Biochemistry and Biophysics, Polish Academy of Sciences, 02-106 Warsaw, Poland;
- 4Institute of Genetics and Biotechnology, Faculty of Biology, University of Warsaw, 02-106 Warsaw, Poland;
- 5International Institute of Molecular and Cell Biology in Warsaw, 02-109 Warsaw, Poland
- Corresponding authors: thj{at}mbg.au.dk; ana.casanal{at}fht.org
Abstract
Biogenesis of most eukaryotic mRNAs involves the addition of an untemplated polyadenosine (pA) tail by the cleavage and polyadenylation machinery. The pA tail, and its exact length, impacts mRNA stability, nuclear export, and translation. To define how polyadenylation is controlled in S. cerevisiae, we have used an in vivo assay capable of assessing nuclear pA tail synthesis, analyzed tail length distributions by direct RNA sequencing, and reconstituted polyadenylation reactions with purified components. This revealed three control mechanisms for pA tail length. First, we found that the pA binding protein (PABP) Nab2p is the primary regulator of pA tail length. Second, when Nab2p is limiting, the nuclear pool of Pab1p, the second major PABP in yeast, controls the process. Third, when both PABPs are absent, the cleavage and polyadenylation factor (CPF) limits pA tail synthesis. Thus, Pab1p and CPF provide fail-safe mechanisms to a primary Nab2p-dependent pathway, thereby preventing uncontrolled polyadenylation and allowing mRNA export and translation.
Keywords
- Nab2p
- Pab1p
- cleavage and polyadenylation factor
- mRNA nuclear export
- poly(A) binding protein
- poly(A) tail length
- polyadenylation
Eukaryotic genes are transcribed into precursor mRNAs (pre-mRNAs) that mature through cotranscriptional processing. The cleavage and polyadenylation (CPA) machinery cleaves and releases the pre-mRNA from the transcribing RNA polymerase II (RNAPII). Subsequent addition of the pA tail to the mRNA 3′ end contributes to the assembly of a messenger ribonucleoprotein complex (mRNP) through the recruitment of PABPs. The PABP-bound pA tail protects the transcript from nuclear degradation and aids in mRNP export (for review, see Oeffinger and Zenklusen 2012; Stewart 2019). Once the mRNP reaches the cytoplasm, the PABP-bound pA tail stimulates translation initiation through its interaction with cap-binding proteins (Brambilla et al. 2019). Finally, shortening of the pA tails by cytoplasmic deadenylases controls mRNA half-life (Eisen et al. 2020). Deadenylation is the first step in the degradation of most mRNAs and is regulated by cis-elements, trans-acting factors, and translation (Parker 2012; Webster et al. 2018). Studies have shown that the lengths of CPA-synthesized pA tails are narrowly distributed and in a species-specific manner (Groner and Phillips 1975; Brown and Sachs 1998; Kühn et al. 2017), ranging from ∼60 to 80 adenosines (As) in S. cerevisiae and Schizosaccharomyces pombe (Lackner et al. 2007; Stewart 2019); to ∼150 As in Danio rerio, Xenopus laevis, and Arabidobsis thaliana; to ∼250 As in humans, Drosophila melanogaster, and Caenorhabditis elegans (Kelly et al. 2014; Subtelny et al. 2014; Azoubel Lima et al. 2017; Kühn et al. 2017; Eisen et al. 2020). The exact molecular mechanisms behind pA tail length control are unclear, but they have been suggested to involve the combined actions of CPA machineries and PABPs (Kühn et al. 2009; Stewart 2019). The pA tail is synthesized by the poly(A) polymerase (Pap1p in S. cerevisiae, and PAP in mammals) subunit of CPA complexes (cleavage and polyadenylation factor [CPF] in S. cerevisiae, and cleavage and polyadenylation specificity factor [CPSF] in mammals). However, CPF/CPSF complexes have distributive polyadenylation activities, and accessory factors are required for efficient polyadenylation (Kühn et al. 2009; Stewart 2019). In mammalian cells, an important accessory factor here is the nuclear PABP PABPN1. In a current model, the first ∼12 As, which are added in a distributive manner, allow binding of PABPN1 to the pA tail and facilitate the transformation of CPSF into a processive enzyme. Successive binding of additional PABPN1 molecules to the growing pA tail occurs until it reaches ∼250 As, after which an elusive structural rearrangement within the pA RNA:PABPN1:CPSF complex is thought to terminate adenosine addition (Kühn et al. 2009). In S. cerevisiae, processive polyadenylation does not appear to require PABPs but is instead dependent on cleavage factor IA (CF IA) (Casañal et al. 2017), a complex consisting of the protein subunits Rna15p, Rna14p, Clp1p, and Pcf11p, which interacts with the “polymerase module” of CPF as well as with the RNA substrate. These interactions are believed to keep the RNA substrate closely tethered to CPF, thereby increasing Pap1p processivity (Casañal et al. 2017).
Although S. cerevisiae PABPs are not required for processive polyadenylation, control of pA tail length still depends on PABP-pA tail association (Stewart 2019). S. cerevisiae harbors two major PABPs, Pab1p and Nab2p, which are essential for cell viability and, during steady state, localize primarily to the cytoplasm and the nucleus, respectively (Brambilla et al. 2019; Fasken et al. 2019). While this localization would intuitively suggest that Nab2p is involved in nuclear pA biogenesis, the high abundance and nuclear–cytoplasmic shuttling behavior of Pab1p (Brune et al. 2005; Dunn et al. 2005) make it an equally relevant candidate. This potential role of Pab1p is in agreement with the fact that Pab1p not only plays a role in controlling cytoplasmic mRNA deadenylation (Parker 2012; Brambilla et al. 2019) but also has been shown to physically interact with the CF IA complex (Amrani et al. 1997; Minvielle-Sebastia et al. 1997). Furthermore, recombinant Pab1p can terminate the polyadenylation reaction in vitro (Minvielle-Sebastia et al. 1997; Hector et al. 2002; Dheur et al. 2005; Viphakone et al. 2008; Schmid et al. 2012), and Δpab1 cells, harboring bypass suppressor mutations that restore viability, exhibit longer pA tails (Caponigro and Parker 1995; Dunn et al. 2005). However, nuclear depletion of Nab2p, or the introduction of point mutations that disrupt its binding to pA tails, also triggers pA tail lengthening in vivo (Kelly et al. 2010; Brockmann et al. 2012; Schmid et al. 2015). Moreover, similar to Pab1p, Nab2p can limit pA tail length in vitro (Hector et al. 2002; Viphakone et al. 2008; Schmid et al. 2012). Therefore, the relative contribution of Nab2p and Pab1p to pA tail length control in vivo remains unknown. Still, it is noteworthy that maximal pA tail lengths only differ modestly upon compromising either Nab2p or Pab1p function (Caponigro and Parker 1995; Brown and Sachs 1998; Hector et al. 2002; Dunn et al. 2005; Brockmann et al. 2012), which suggests functional redundancy or the involvement of additional mechanisms.
A long-standing challenge in determining nuclear pA tail lengths in vivo has been that deadenylation ensues as soon as the polyadenylated transcript enters the cytoplasm, which obscures any information about the initial length control process. Additionally, cytoplasmic and nuclear constituents are readily mixed in cellular extract preparations used for in vitro polyadenylation reactions, rendering ex cellulo experiments inconclusive. Here, we circumvent these problems by using two complementary approaches. First, we effectively uncouple nuclear polyadenylation from nuclear export—and, consequently, cytoplasmic deadenylation—to study the length determinants of newly synthesized pA tails. Second, we reconstitute the polyadenylation reaction in vitro using recombinant components. Together, our results suggest that Nab2p, Pab1p, and the CPF-cleavage factor complex exercise a three-layered control of nuclear pA tail length.
Results
Preventing mRNA export reveals a major role for Nab2p in nuclear pA tail length control
Analysis of nuclear RNA pA tail biogenesis is confounded by the opposing process of cytoplasmic deadenylation. To circumvent this, we used the anchor-away (AA) system (Haruki et al. 2008) to deplete the main mRNA export factor Mex67p from yeast nuclei (Tudek et al. 2018), thereby facilitating examination of newly formed pA tails before they encounter cytoplasmic deadenylases. To focus our analysis on transcripts produced during the nuclear export block, the expression of heat shock RNAs (hsRNAs) was induced by shifting MEX67-AA cultures from 25°C to 38°C 5 min after rapamycin-stimulated depletion of nuclear Mex67p (Fig. 1A). High-resolution Northern blotting analysis of RNA from control cells, subjected to a 15-min heat shock, revealed a strong increase in the levels of HSP104 transcripts with pA tails ranging between zero and ∼60 As (Fig. 1B, cf. lanes 1 and 5; Supplemental Fig. S1A, left panel, cf. lanes 5 and 6), suggesting that many of these transcripts had undergone cytoplasmic deadenylation. Nuclear depletion of Mex67p caused the disappearance of a large fraction of HSP104 transcripts, and consistent with previous observations (Hilleren and Parker 2001; Jensen et al. 2001; Tudek et al. 2018), the remaining RNAs were either unadenylated or carried longer “hyperadenylated” pA tails of >70 As (Fig. 1B, lanes 2–4,6). HSP104 RNAs were nuclear-confined in rapamycin-treated cells, whereas in untreated cells, HSP104 transcripts were localized throughout the nucleus and cytoplasm (Supplemental Fig. S1B, top row panels). Collectively, these results indicated that regular nuclear pA tail length control was compromised when Mex67p was depleted from the nucleus.
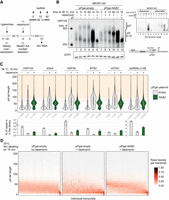
Nab2p controls nuclear mRNA pA tail length. (A) Experimental scheme to measure nuclear mRNA pA tail lengths. mRNA export was conditionally blocked in MEX67-AA cells, overexpressing Nab2p or not, 5 min before transferring cells to 38°C (dark line on top) or initiating 4tU labeling (dashed dark line). RNA samples were collected at the indicated times after heat treatment or initiation of 4tU labeling. (B) RNase H/Northern analysis of HSP104 RNA 3′ ends from MEX67-AA cells subjected to different 38°C incubation periods, conditionally depleted of nuclear Mex67p (rapamycin −/+) and expressing endogenous levels of Nab2p (“pPgal-empty”) or overexpressing Nab2p from a plasmid under control of a galactose inducible promoter (“pPgal-NAB2”) as indicated. (Bottom right panel) HSP104 RNA was detected using an oligonucleotide probe close to the pA site and, to increase resolution, transcripts were internally cleaved with RNase H targeted by the DL163 DNA antisense oligo, annealing 230 nt upstream of the pA site. A subset of RNase H treatments further included a dT18 oligo (dT −/+) to trim away pA tails. SCR1 RNA was probed as a loading control. Migration of HSP104 RNA harboring normal (pA+, 0–70 As) and hyperadenylated (pA++, >70 As) pA tails are indicated. Pixel intensities have been linearly enhanced to emphasize the hyperadenylated RNA. The top right panel inset shows the same exposure in full pixel intensity range. (C) Quantification of hsRNA pA tail lengths by direct RNA sequencing. Polyadenylated RNA was extracted from MEX67-AA cells shifted for 15 min to 38°C without (“−”) or with (“+”) a 5-min pretreatment with rapamycin, and without (gray shaded violins) or with (green violins) ectopic Nab2p expression as in B. pA tail lengths are shown for five individual hsRNAs and a meta-analysis of the top 48 most heat-induced mRNAs. pA tails >70 As (orange-shaded area above the red dashed line) were defined as hyperadenylated. The number of sequence reads in each group is denoted below the violins. Bar plots below the graphs display the mean percentage of reads from shown transcripts within the sequenced DRS libraries. Individual values from two independent replicates are shown as dots. (D) pA tail length distributions of 4tU RNAs. Reads from each RNA were grouped based on their pA tail lengths in 10-A bins, and bin values were then normalized to the total number of reads for each transcript. The resulting fraction of reads in each bin is represented using the indicated color gradient. Only transcripts containing >20 reads in the “pPgal-NAB2 + rapamycin” sample were included in the analysis (n = 821). Transcripts were sorted by their median pA tail length in the right panel.
Our earlier work showed that reduced levels of newly synthesized mRNA, following Mex67p nuclear depletion, result from their destabilization due to decreased availability of Nab2p, which becomes sequestered on the accumulating nuclear pA RNAs (Tudek et al. 2018). Consistently, nuclear depletion of Nab2p caused a similar hyperadenylation of HSP104 RNA (Schmid et al. 2015). Moreover, nuclear codepletion of Mex67p and Nab2p did not exacerbate the observed hyperadenylation phenotype (Supplemental Fig. S1C, cf. lanes 2 and 3), suggesting that the individual depletions impact the same cellular process. Based on these considerations, we hypothesized that the failure to control pA tail length in Mex67p-depleted nuclei was caused by the insufficient availability of Nab2p. Indeed, sixfold overexpression of Nab2p, under Mex67p depletion conditions (Supplemental Fig. S1A, right panel; Supplemental Fig. S1B), partially recovered HSP104 transcript levels and reduced their pA tail lengths (Fig. 1B, lanes 8–10; Supplemental Fig. S1A, lanes 1–4). Such “Nab2p-induced” pA tail length of ∼50–60 As matched the upper length of HSP104 RNA pA tails in cells supporting normal mRNA biogenesis (Fig. 1B, cf. lane 5 and lanes 9,10; Supplemental Fig. S1A, cf. lanes 3 and 5), demonstrating a direct role of Nab2p in nuclear HSP104 RNA pA tail length control.
To interrogate Nab2p-dependent nuclear pA tail length control on a wider set of heat-induced transcripts, we next subjected polyadenylated RNA from these samples to Oxford Nanopore Technologies direct RNA sequencing (DRS), estimating pA tail lengths of individual mRNAs using the Nanopolish software (Workman et al. 2019; Tudek et al. 2021). We identified 48 hsRNAs whose expression was up-regulated greater than threefold upon heat treatment (Supplemental Table S2). Again, nuclear depletion of Mex67p resulted in increases of both the mean pA tail lengths and the fractions of hyperadenylated tails, which was evident for individual transcripts and in a combined analysis of the 48 hsRNAs (Fig. 1C, gray violin plots). Note that DRS RNA pA tail length estimates display considerable variance, especially for long pA tails (Krause et al. 2019). Mex67p depletion had largely no effect or led to pA tail shortening on noninduced RNAs, presumably because their pA profiles were dominated by transcripts already transported to the cytoplasm prior to Mex67p depletion and deadenylated outside the nucleus during the export block (Supplemental Fig. S1D, violins 13,14,17,18). Nonetheless, even among genes whose expression was not strongly affected by heat exposure, a few highly expressed transcripts were identified that displayed hyperadenylation in response to Mex67p depletion (Supplemental Fig. S1D, gray violins for HHF1 and TMA10), demonstrating that disruption of nuclear pA tail length control was not restricted to bona fide hsRNAs. Notably, all instances of Mex67p depletion-induced hyperadenylation were largely suppressed by Nab2p overexpression, decreasing pA tail lengths to ∼60 As (Fig. 1C; Supplemental Fig. S1D). Corrected tail lengths were generally accompanied by increased mRNA levels (Fig. 1C, bottom row bar graphs). As a corollary, we noticed that Nab2p overexpression in control cells tended to favor pA tail lengths to be enriched at ∼60 As (Fig. 1B, cf. lanes 5 and 11; Supplemental Fig. S1D, green violins without rapamycin). We attribute this to possible interference of excess cellular Nab2p with Pab1p-mediated deadenylation in the cytoplasm since Nab2p overexpression did not affect the mainly cytoplasmic localization of HSP104 RNAs in control cells (Supplemental Fig. S1B, cf. panels 4 and 9).
To further interrogate pA tail lengths of non-hsRNAs, we metabolically labeled MEX67-AA cells, grown at 25°C, with 4-thio-uracil (4tU) for 15 min and DRS-sequenced the isolated 4tU-incorporated RNAs (Fig. 1A). These contained longer pA tails, ranging down from ∼70 As, as compared with those of a control sample not subjected to 4tU (Supplemental Fig. S1E, cf. violins 1 and 2). pA tail length distributions of individual transcripts showed similar maximal lengths across the majority of the 821 examined transcripts (Fig. 1D, left panel). Nuclear pA tails were again inspected by depleting Mex67p from the nucleus with rapamycin for 5 min before the start of the 4tU labeling. This resulted in two opposing effects: pA tails generally underwent shortening while hyperadenylated transcripts became relatively more frequent (Fig. 1D, cf. left and middle panels), which was exemplified by the often-studied model mRNA CYC1 pA tail distribution, where the fraction of both short and long tails increased upon nuclear Mex67p depletion (Supplemental Fig. S1E, violins 7,8). We suspect that the reduced bulk pA tail length can be attributed to the rapid degradation of mRNAs that are confined to the nucleus (Tudek et al. 2018). On the other hand, the increased hyperadenylation indicated a general defect in pA tail length control, which Nab2p overexpression was again able to counteract (Fig. 1D; right panel; Supplemental Fig. S1E).
All of our results together suggest that Nab2p directly controls the length of nuclear pA tails in S. cerevisiae.
Sustained control of nuclear pA tail length in the absence of Nab2p
Like Nab2p, Pab1p has been linked to nuclear pA tail length control (Amrani et al. 1997; Minvielle-Sebastia et al. 1997; Schmid et al. 2012), although its in vivo relevance has been questioned (Hector et al. 2002; Dheur et al. 2005; Viphakone et al. 2008). To investigate this issue using our in vivo assay, we used a mutant variant of Pab1p that is not imported into the nucleus due to deletion of its nuclear localization signal (NLS)-containing RRM4 domain (Brune et al. 2005; Dunn et al. 2005). The mutant protein maintains its RNA and eIF4G binding capacities and supports pA-dependent translation (Kessler and Sachs 1998), and the growth of cells expressing pab1ΔRRM4 as their sole copy of Pab1p was only marginally impaired (Supplemental Fig. S2A). In the absence of rapamycin, MEX67-AA/pab1ΔRRM4 cells displayed slightly longer HSP104 pA tails, possibly due to slowed cytoplasmic deadenylation. Still, the maximal pA tail length remained largely unchanged (Fig. 2A, cf. lanes 2 and 4). Further analysis by DRS recapitulated this finding (Fig. 2B; Supplemental Fig. S2B); i.e., the fraction of the transcripts examined carrying pA tails >100 As remained low in MEX67-AA/pab1ΔRRM4 cells (Supplemental Fig. S2C, cf. bars 3 and 9). This implied that Nab2p normally limits nuclear pA tail synthesis, and Pab1p has little effect on nuclear pA tail length control when Nab2p is available.
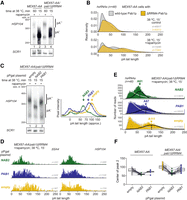
Nab2p-independent mechanisms for terminating nuclear polyadenylation. (A) RNase H/Northern blot analysis of HSP104 RNA 3′ ends as in Figure 1B, but for MEX67-AA and MEX67-AA/pab1ΔRRM4 cells with or without nuclear depletion of Mex67p as indicated. (B) DRS-derived pA tail length distributions of hsRNAs (n = 48) from MEX67-AA (gray density) or MEX67-AA/pab1ΔRRM4 (yellow density) cells. Individual transcript examples are shown in Supplemental Figure S2B. (C) RNase H/Northern analysis of HSP104 RNA 3′ ends as in Figure 1B, but for MEX67-AA/pab1ΔRRM4 cells expressing endogenous levels of PABPs (pPgal “empty”) or overexpressing Nab2p or Pab1p from plasmid-borne copies (pPgal-NAB2 or pPgal-PAB1) (Supplemental Fig. S2D). Phosphorimager lane scans are shown at the right with arrows marking the main RNA pA tail length peaks of different samples. (D,E) DRS-derived pA tail length distributions of SSA4 and HSP104 RNAs (D) or of hsRNAs (n = 48; E) displaying individual reads for the samples described in C, with bimodal distributions fitted as detailed in Supplemental Figure S2F. Peak centers of the long pA tail distributions (black lines) are reported. Note that the larger variance of the DRS estimates for the long pA tails flatten their distribution compared with the short pA tails. (F) Quantification of DRS long pA tail distribution centers among individual heat-induced transcripts (connected dots) in rapamycin-treated samples. Box plot displays mean ± SD of plotted values.
As expected, rapamycin treatment of MEX67-AA/pab1ΔRRM4 cells resulted in the hyperadenylation of HSP104 RNA (Fig. 2A, cf. lanes 3 and 4). Interestingly, closer inspection of the Northern blot revealed that these tails were slightly longer than those of HSP104 RNAs in cells expressing WT Pab1p (Fig. 2A, cf. lanes 1 and 3). Consistently, pA tails of hsRNAs, as measured by DRS, displayed longer tails (Fig. 2B; Supplemental Fig. S2B) with an approximately twofold increase in the fraction of transcripts with pA tails >100 As in MEX67-AA/pab1ΔRRM4 compared with MEX67-AA cells (Supplemental Fig. S2C). Taken together, this indicates that Pab1p can restrict nuclear pA tail length, and this can be observed when Nab2p is not available.
To corroborate this idea, we overexpressed WT Pab1p in Mex67p nuclear-depleted cells (Supplemental Fig. S2D), which resulted in the shortening of the hyperadenylated HSP104 RNA pA tails in both MEX67-AA/pab1ΔRRM4 (Fig. 2C, cf. lanes 1 and 3) and MEX67-AA (Supplemental Fig. S2E, cf. lanes 1 and 3) cells. However, these tails, presumably controlled by Pab1p, were still noticeably longer than their Nab2p-controlled counterparts (Fig. 2C, cf. lanes 2 and 3; Supplemental Fig. S2E cf. lanes 2 and 3, and lanes 5 and 6). This was in agreement with the DRS reads of individual and pooled hsRNAs (Fig. 2D,E). To focus our analysis of DRS reads on the newly synthesized RNA fraction, we fitted the pA tail distributions to a bimodal model (Supplemental Fig. S2F; see Supplemental Material). This suggested that the de novo lengths of long pA tails in cells overexpressing Nab2p and Pab1p were 61 As and 87 As, respectively (Fig. 2E,F). These are both clearly shorter than the 111 As in cells without PABP overexpression, and it therefore appears that Pab1p can control nuclear pA tails and that this activity partially limits pA tail lengths in Mex67p nuclear-depleted cells. Importantly, such Pab1p-dependent tail length control is only exposed when nuclear levels of Nab2p are limiting. A further noteworthy aspect of these results is that depletion of Mex67p in pab1ΔRRM4 cells, where neither Nab2p nor Pab1p would be available to control nuclear pA tail length, still created a situation with controlled polyadenylation; i.e., hyperadenylated HSP104 RNA pA tails exhibited a rather narrow length distribution of 100–150 As (Fig. 2C, lane 1, note quantification at the right of the gel image), which was also evident in the DRS data of hsRNAs (Fig. 2E,F). This suggests that an additional and PABP-independent mechanism is in place to limit nuclear pA tail length.
Nab2p and Pab1p control CPF-mediated polyadenylation in vitro but with distinct properties
Having established that three independent mechanisms limit pA tail length in vivo, we tested whether they could also be reconstituted in vitro. For this, we expressed and purified a 14-subunit recombinant S. cerevisiae CPF complex, as well as CF IA, CF IB, Nab2p, and Pab1p (Fig. 3A; Kumar et al. 2021). To perform polyadenylation assays with the recombinant components, we used a 5′-FAM-labeled, 120-nt synthetic pre-mRNA, consisting of the 3′ UTR of the CYC1 gene ending at the cleavage site (pcCYC1). CPF, CF IA, and CF IB were incubated with pcCYC1 and Nab2p in a time-course polyadenylation experiment, showing that ∼60 As were added onto the RNA (Fig. 3B, left). Such pA tail length control, comparable with the one previously seen in vivo, was observed at Nab2p concentrations of ≥200 nM, two times the concentration of RNA in the assay (Supplemental Fig. S3A). This is consistent with a requirement for a Nab2p dimer to bind each RNA molecule (Aibara et al. 2017). In contrast, incubation with the CPF complex alone resulted in pA tails of >1000 nt (Fig. 3B, right). Similar results were obtained on a 180-nt pcCYC1 pre-mRNA 3′ UTR using endogenous CPF (native CPF) purified from yeast (Supplemental Fig. S3B), validating the recombinant CPF complex activity. In the absence of any cleavage factors, Nab2p inhibited CPF-mediated polyadenylation (Supplemental Fig. S3C). This may be a consequence of nonspecific RNA binding by Nab2p that blocks CPF access to the RNA substrate and that is normally outcompeted by specific (presumably higher affinity) RNA binding by CF IA and/or CF IB. Taken together, these in vitro assays showed that Nab2p limits the addition of adenosines to ∼60 As during RNA 3′ end polyadenylation in a reconstituted system.
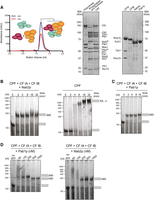
Nab2p and Pab1p control pA tail length in a reconstituted polyadenylation system. (A, left) Size exclusion chromatogram of recombinant CPF reconstitution from the polymerase module (mint) and combined phosphatase (orange) and nuclease (magenta) modules. Tagged proteins are indicated with a star. (Right) Coomassie-stained SDS-PAGE analysis of purified proteins and complexes used for the polyadenylation assays. (B–D) Denaturing RNA gels of polyadenylation assays using a fluorescently labeled 120-nt pcCYC1 RNA substrate (final concentration 100 nM). Adenylated and nonadenylated forms of the substrate are indicated as gray rectangles. All assays were repeated at least three times, and representative gels are shown. (B) Time course analysis of polyadenylation by CPF in the presence or absence of Nab2p (600 nM) and cleavage factors CF IA and CF IB, as indicated above the gel panels. (C) Time-course analysis of polyadenylation by CPF in the presence of Pab1p (1 µM) and the cleavage factors CF IA and CF IB. (D) Effect of Pab1p (left panel) and Nab2p (right panel) concentrations on CPF/CF IA/CF IB-synthesized pA tail lengths. Polyadenylation reactions were initiated by the addition of ATP and stopped after 8 min. First gel lanes at the left show reactions in the absence of ATP. Time courses for corresponding reactions are displayed in Supplemental Figure S3, A and D.
We next investigated whether Pab1p might also limit polyadenylation in vitro. Indeed, in the presence of Pab1p, the CPF/CF IA/CF IB-mediated reaction resulted in the addition of ∼60 As to the substrate RNA (Fig. 3C). As the Pab1p-controlled pA tails were slightly shorter than observed in vivo, we investigated the concentration dependence of this Pab1p activity. This included Pab1p concentrations that were both sub- and superstoichiometric compared with the 100 nM concentration of the RNA substrate. Notably, at 60 or 250 nM Pab1p, pA tails of ∼90 As were added to the substrate (Fig. 3D; Supplemental Fig. S3D), while at higher Pab1p concentrations (500 nM or 1 µM), the pA tails were distinctively shorter and of a similar length to those observed in the presence of Nab2p (Fig. 3D, cf. Pab1p and Nab2p panels). Similar results were obtained using native CPF (Supplemental Fig. S3E). Thus, Pab1p limits the length of the pA tails synthesized by CPF/CF IA/CF IB in vitro, and the exact length is dependent on the Pab1p concentration. The concentration dependence can be reconciled as a competition between polyadenylation and Pab1p association, with higher Pab1p concentration favoring earlier association and termination of polyadenylation. Notably, the Pab1p-controlled pA tails are ∼60 or ∼90 As, consistent with two or three Pab1p molecules bound to pA (Baer and Kornberg 1980).
In summary, both Nab2p and Pab1p control pA tail length in vitro in a similar manner to their roles in vivo.
CPF with CF IA and CF IB harbors an intrinsic capacity to control pA tail length
As pA tail lengths remained restricted in vivo even in the absence of both Nab2p and Pab1p (Fig. 2), we also performed in vitro CPF/CF IA/CF IB-mediated polyadenylation assays in the absence of any PABPs. Surprisingly, these assays showed that polyadenylation ceased after the addition of ∼100–300 As to the substrate RNA (Fig. 4A). This was in stark contrast to the very long tails (>1000 As) synthesized by CPF in the absence of CF IA and CF IB (Fig. 3B), or shorter tails synthesized when either Nab2p or Pab1p was present (Fig. 4B). Similar results were observed when polyadenylation was coupled to the cleavage of a 56-nt CYC1 pre-mRNA substrate (Supplemental Fig. S4A) or when polyadenylation assays were carried out with the native CPF complex and pcCYC1–180 nt (Supplemental Fig. S4B).
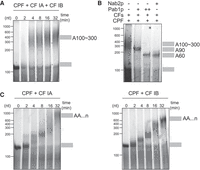
CPF/CF IA/CF IB harbors an intrinsic mechanism for controlling pA tail length. (A) Time-course analysis of polyadenylation as in Figure 3B, but by CPF in the presence of both cleavage factors CF IA and CF IB. (B) CPF-synthesized pA tails in the presence of CF IA and CF IB (CFs) and either low (“+”; 250 nM) or high (“++”; 1 µM) concentrations of Pab1p or Nab2p (600 nM). Assays were performed as in Figure 3D. (C) Time-course analysis of polyadenylation by CPF in the presence of either CF IA (left panel) or CF IB (right panel).
It therefore appeared that CPF/CF IA/CF IB possesses an inherent capacity to restrict pA tail length in vitro, in the absence of other cellular factors. To determine which components contribute to such “intrinsic” pA tail length control, we performed polyadenylation assays in the presence of only one of each of the cleavage factors. In the presence of CF IA alone, CPF initially added pA tails of 70–120 As, which over time were elongated to >1000 As (Fig. 4C, left panel). In the presence of only CF IB, CPF added several hundreds of As (Fig. 4C, right panel). In both cases, the RNA substrate was polyadenylated more slowly than when CPF was assembled with both CF IA and CF IB. We therefore conclude that the fully assembled 3′ end machinery, including CPF, CF IA, and CF IB, contains an alternative mechanism for pA tail length control, which is intrinsic to the complex. More generally, our in vitro and in vivo data together show that three different pathways control nuclear pA tail length: Nab2p, Pab1p, and CPF/CF IA/CF IB.
PABP-independent termination of pA tail synthesis produces functional mRNA
Given the role of the pA tail and PABPs in mRNA export and translation (Brambilla et al. 2019; Stewart 2019), we wanted to determine whether functional mRNAs can be produced by all three mechanisms for terminating pA tail synthesis. To address this question, we treated cells with [35S]-methionine to label and detect newly synthesized proteins before and after heat shock (Stutz et al. 1997; Kallehauge et al. 2012). As expected, heat shock proteins (Hsp104p, Hsp82p, Hsp70p) were translated in heat-induced MEX67-AA cells but were not detectable in cells grown at 25°C or if mRNA export was blocked by the addition of rapamycin (Fig. 5A, lanes 1–5; see full gel image in Supplemental Fig. S5A). We then used the AA-system to deplete Nab2p from the nucleus (Supplemental Fig. S5B; Schmid et al. 2015) so that polyadenylation would be terminated solely by the Pab1p-dependent and/or CPF/CF IA/CF IB-dependent mechanisms. Notably, heat shock proteins were produced in cells depleted of nuclear Nab2p, although at lower levels (Fig. 5A, lanes 6–10). This ∼50% reduction of Hsp104p production mirrored the decrease of HSP104 mRNA levels in this condition (Fig. 5B,C; Supplemental Fig. S5C; Schmid et al. 2015; Tudek et al. 2018). Examination of HSP104 mRNA pA tails verified the loss of normal pA tail length control (Fig. 5C; Supplemental Fig. S5C), demonstrating that Nab2p was absent at the time of termination of pA tail synthesis of these transcripts and making it unlikely that residual nuclear Nab2p would (solely) explain heat shock protein production in this condition. Thus, we conclude that mRNPs resulting from Nab2p-independent pA tail termination are export- and translation-competent.
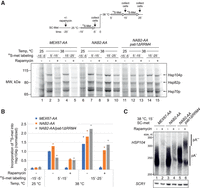
hsRNAs expressed in the absence of nuclear PABPs still direct protein synthesis. (A) [35S]-Met protein labeling analysis of the indicated cell cultures (SDS-PAGE; bottom) following the experimental scheme displayed at the top. Rapamycin, or DMSO (−rapamycin), was added 20 min before heat induction, followed by [35S]-Met labeling for one of three indicated time periods (gray horizontal arrows): (1) at 25°C for 5–15 min before the temperature shift (−15′–5′), or (2) for 5–15 min (5′–15′) or (3) for 15–25 min (15′–25′) after the 38°C shift. Markers for heat shock proteins are indicated on the right. (B) Quantification of Hsp104p expression from A. Incorporation of [35S]-Met into Hsp104p in each gel lane was normalized to the total [35S]-Met signal from proteins <50 kDa (see Supplemental Fig. S5A). Bars display averaged values normalized to the “MEX67-AA, − rapamycin, 38°C 5′–15′” samples. Individual values from two independent replicates are shown as dots. (C) RNase H/Northern blotting analysis of HSP104 mRNA 3′ ends as in Figure 1B, but in conditions used for [35S]-Met protein labeling. RNA samples were from cells harvested 15 min after transfer to 38°C. Additional time points are shown in Supplemental Figure S5C. Note the loss of HSP104 RNA signal and the appearance of hyperadenylated transcripts following nuclear depletions of Mex67p and Nab2p.
This raised the question of whether nuclear Pab1p and CPF/CF IA/CF IB can both compensate for the absence of Nab2p. To address this issue, we constructed NAB2-AA cells expressing as their sole source of Pab1p the cytoplasmic ΔRRM4 mutant variant. Again, robust heat shock protein production was detected both in the absence and presence of rapamycin, showing that functional mRNAs are produced in the absence of both Nab2p and Pab1p in the nucleus (Fig. 5A, lanes 11–15). Notably, pab1ΔRRM4 cells displayed a delay in heat shock protein production (Fig. 5B), in line with the proposed role of Pab1p in mRNA biogenesis, export, and translation (Caponigro and Parker 1995; Brune et al. 2005; Dunn et al. 2005; Brambilla et al. 2019). Interestingly, the majority of detectable HSP104 mRNA remained hyperadenylated despite ongoing export (Fig. 5C, lane 6), which is at least partially explained by the cytoplasmic deadenylation defect in pab1ΔRRM4 cells (Supplemental Fig. S5C). We obtained similar results after depletion of nuclear Nab2p in a Δpab1 strain containing the spb2-1 bypass suppressor mutation that restores the viability of the PAB1 deletion (Supplemental Fig. S5D–F; Brambilla et al. 2019). We conclude that export- and translation-proficient mRNA can be produced in the absence of nuclear PABPs despite a marked decrease in mRNA levels and pA tail lengthening.
Discussion
The exact requirements for nuclear pA tail length control have remained elusive. Data presented here suggest that, in yeast, nuclear polyadenylation carried out by the CPF complex can be terminated by at least three distinct mechanisms, each giving rise to a different length of de novo synthesized pA tails (Fig. 6). Nab2p is the main regulator of polyadenylation, controlling the length of pA tails to ∼60 As. When Nab2p levels are insufficient, two fail-safe mechanisms, possibly acting in parallel, prevent uncontrolled polyadenylation. The first relies on the nuclear pool of Pab1p and results in pA tails of 60–90 As in length. The second mechanism is carried out by the CPF-cleavage factor complex terminating pA tail synthesis after the addition of 100–200 As.
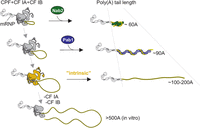
Model: three-layered control of pA tail length in S. cerevisiae. Polyadenylation carried out by the CPF/CF IA/CF IB complex (dark gray) is primarily terminated by Nab2p (green), whose association with the mRNP (dark line and light-gray shapes depicting proteins and RNA, respectively) produces ∼60-A-long pA tails (light yellow). If Nab2p is not available, either the nuclear pool of Pab1p (blue) or the CPF complex, together with the cleavage factors CF IA and CF IB (highlighted in dark yellow at this step), provides a fail-safe termination of polyadenylation, producing longer pA tails.
Nuclear pA tail length is primarily controlled by Nab2p and backed by Pab1p
The in vivo Nab2p-dependent pA tails of ∼60 As are also observed when Nab2p is added to CPF/CF IA/CF IB-directed in vitro polyadenylation reactions, indicating that Nab2p alone is sufficient to terminate polyadenylation by the CPF. This is in line with previous in vitro studies (Viphakone et al. 2008) and with the finding that Nab2p restricts bulk pA tails in vivo (Hector et al. 2002; Kelly et al. 2010).
Our results showing Pab1p-dependent termination of polyadenylation are also consistent with previous studies (Brown and Sachs 1998; Dunn et al. 2005; Viphakone et al. 2008). However, Pab1p mediates the production of ∼90-nt pA tails, which are longer than the maximal pA tails in unperturbed cells, indicating that Pab1p does not normally control this process. Moreover, we found that impairing Pab1p function inhibits pA tail deadenylation globally, which is in line with a major role for Pab1p in the stimulation of cytoplasmic deadenylation activities (Webster et al. 2018; Yi et al. 2018; Brambilla et al. 2019; Schäfer et al. 2019). Together, these data argue that Nab2p and Pab1p play distinct primary roles in controlling pA tail lengths in the nuclear and cytoplasmic stages of the mRNA life cycle, respectively. This division of activities is consistent with the steady-state localization of Nab2p and Pab1p, and with the model that active remodeling of mRNPs at the cytoplasmic face of the nuclear pore complex facilitates the exchange of Nab2p with Pab1p (Tran et al. 2007). Still, the nuclear–cytoplasmic shuttling behavior of Pab1p will allow the protein to compensate for any imbalances in nuclear Nab2p levels, providing an auxiliary fail-safe mechanism.
Estimated concentrations of Nab2p and mRNA in S. cerevisiae nuclei are within the same range (∼2.8 µM and ∼0.7–2.9 µM, respectively) (see the Supplemental Material). Furthermore, a major share of nuclear polyadenylated RNA is expected to be associated with Nab2p, leaving the freely available pool of Nab2p substantially smaller. This supports the idea that fluctuations in the rates of mRNA production and trafficking may lead to a temporary depletion of soluble Nab2p, which would warrant buffering by fail-safe termination mechanisms. Moreover, instances of aberrant Nab2p localization, or availability, have been reported during severe heat stress (Carmody et al. 2010) or when nuclear RNA processing pathways are disrupted (Paul and Montpetit 2016; Aguilar et al. 2020). Interestingly, phenotypes associated with limiting Nab2p levels are partially restored by overexpression of Pab1p (Hector et al. 2002; Aguilar et al. 2020), lending further support to the idea that Pab1p and Nab2p have redundant nuclear functions. On the other hand, decreasing cytoplasmic mRNA abundance has been shown to drive nuclear localization of mammalian PABPC (Kumar et al. 2011; Gilbertson et al. 2018), so it is possible that global changes in RNA metabolism determine which pA termination pathway is primarily used. Finally, our data do not rule out the possibility that Pab1p could be recruited to specific transcripts in the nucleus to affect their processing.
Since similar pA tail lengths are observed in vivo and in vitro, we suggest that PABPs determine tail lengths by terminating the polyadenylation process. The finding that two nonhomologous PABPs can control pA tail length implies that the mere association of a PABP with the pA tail suffices to terminate CPF-instructed polyadenylation. How the association of Nab2p or Pab1p to pA tails terminates polyadenylation and how PABPs “measure” the pA tail length are important topics of future research and likely depend on the formation of multimeric PABP assemblies on the pA tail (Aibara et al. 2017; Schäfer et al. 2019).
Intrinsic pA tail length control by the CPF complex
Our data also revealed a surprising PABP-independent mechanism of nuclear pA tail length control. While we cannot formally exclude that other PABP-like protein(s) affect the polyadenylation reaction in vivo, our demonstration that such length control can be reconstituted with fully purified components strongly argues that the CPF complex with CF IA and CF IB harbors a built-in activity to limit pA tail length. Moreover, this capacity of CPF appears to be relevant in vivo when nuclear PABP levels are limiting. What might be the mechanism underlying such pA tail length control? Efficient polyadenylation requires the maintenance of tripartite interactions between the RNA-binding subunits of CPF, Pap1p, and RNA (Zhelkovsky et al. 1998; Ezeokonkwo et al. 2011). Disruption of any of these interactions would be expected to reduce the efficiency of polyadenylation and facilitate termination. As cleavage factors interact directly with the substrate RNA and the CPF complex (Ezeokonkwo et al. 2011; Holbein et al. 2011; Casañal et al. 2017), they likely help to maintain CPF:substrate interactions in an active polyadenylating complex. In the absence of any PABPs, the interaction of the cleavage factors and the product RNA may sterically disfavor a long pA tail from accessing the active site of Pap1p and thereby lead to termination. Such a model would explain how cleavage factors can regulate the polyadenylation activity of CPF. Another possibility is that the pA tail competes for binding to CPF or CF IB (Hill et al. 2019), displacing them from the 3′ UTR sequences. We can also speculate that intrinsic length control represents a case of product inhibition on Pap1p in which the product (the pA tail) acts in cis to inhibit its own polymerization in a length-dependent manner.
Interestingly, similar to this study in yeast, nuclear pA tails converge to a length of 100–200 As when nuclear PABP is depleted in human cells (Kühn et al. 2017), which indicates a potentially conserved pA tail length control mechanism in eukaryotes. We anticipate that pA tail synthesis is controlled by multiple pathways also in other organisms.
Fail-safe termination of polyadenylation
Efficient nuclear export relies on proper mRNA 3′ end processing (for review, see Oeffinger and Zenklusen 2012; Stewart 2019). One aspect of this connection is exemplified by Nab2p, which binds to polyadenylated mRNA as well as to the nuclear export factor Mex67p (Hector et al. 2002; Stewart 2019). Nab2p binding not only impacts the pA tail length of the mRNA but is also required for its nuclear stability (Schmid et al. 2015; Tudek et al. 2018). Tight coupling of mRNA 3′ end processing and nuclear export may therefore facilitate a one-way system for rapid delivery of Nab2p-associated transcripts with ∼60-nt-long pA tails to the cytoplasm. However, a considerable portion of transcripts is still being exported and translated in Nab2p-depleted cells, indicating that the Pab1p-dependent and CPF-cleavage factor complex-dependent “fail-safe” mechanisms also facilitate gene expression. How might 3′ end processing and export cooperate in these cases? Nuclear export of Pab1p can be mediated by its direct interaction with Crm1p (Brune et al. 2005; Dunn et al. 2005), but clearly this pathway does not provide an alternative export route for hsRNAs in Mex67p-depleted cells. Thus, fail-safe-terminated transcripts are likely to be exported in a Mex67p-dependent manner, and Pab1p association might serve to protect them against degradation. It is less obvious how CPF-cleavage factor complex-terminated transcripts may evade degradation, but their hyperadenylated pA tails could potentially equip them with a longer survival time in the nucleus.
Before a polyadenylated transcript is able to exit the nucleus, it must detach from the CPF complex. It has been speculated that the association of PABPs would trigger this release step (Dunn et al. 2005; Fasken et al. 2019; Stewart 2019). Our observation that mRNAs can be exported and translated in cells with nuclear-depleted PABPs indicates that such release can also be achieved by other factors, although PABPs may, of course, speed up the process. Nonetheless, termination of polyadenylation must precede or coincide with the release of RNA from the CPF:substrate complex (Qu et al. 2009; Stewart 2019), suggesting that fail-safe termination mechanisms will allow transcript release and facilitate the recycling of the polyadenylation machinery to new RNA substrates. In this way, a three-layered control of the polyadenylation process ensures that the production of mRNA and its export to the cytoplasm does not come to a halt if one factor is temporarily unavailable. Overall, this mechanism will help to ensure the robustness of gene expression.
Materials and methods
Yeast strains and cultures
Yeast strains and plasmids are listed in Supplemental Tables S3 and S4. For most experiments, cells were cultured in synthetic complete minimal medium lacking uracil and containing 2% raffinose and 0.1% glucose (SC-ura). Three hours before heat shock, 2% galactose was added to the cell cultures, which had an optical density (OD600) ∼0.5 to induce the protein expression from pPgal plasmids. For heat shock experiments, cultures kept at 25°C were heat-shocked by rapidly mixing an equal volume of media preheated to 51°C and transferring the cultures to a 38°C water bath. In metabolic labeling experiments, cultures were maintained at 25°C throughout the 4tU-labeling period (final 100 µM; ACROS Organics). Rapamycin (final 1 µg/mL; Life Technologies) was added to cultures before heat shock or addition of 4tU at time points specified in the figures. Cells for RNA analysis were harvested by mixing equal volumes of culture with ethanol precooled on dry ice and centrifuging at 3000g for 3 min at 4°C. Pellets were washed twice with ice-cold water and stored at −80°C. Endogenous CPF was purified from Ref2-TAPS strain grown at 30°C in YPD media and harvested at an OD600 of 6–7.
RNase H Northern blotting
Twenty micrograms of total RNA, extracted with the hot phenol method detailed in the Supplemental Material, was combined with 2 µM RNase H targeting oligonucleotides (sequences are listed in Supplemental Table S1) in annealing buffer (50 mM Tris-HCl at pH 8.3, 50 mM KCl) in a total volume of 12 µL. The mix was incubated for 2 min at 85°C and slowly cooled to 37°C. Subsequently, 8 µL of a mix preheated to 37°C and containing 2.5 U of RNase H (New England Biolabs), 2.5× RNase H reaction buffer (1× buffer: 50 mM Tris-HCl, 75 mM KCl, 3 mM MgCl2, 10 mM DTT at pH 8.3), 25 mM DTT, and 4U RiboLock RNase inhibitor (Thermo Scientific) was added and incubated for 30 min at 37°C. Next, RNA was precipitated by the addition of 100 µL of absolute ethanol and 20 µL of solution containing 600 mM NaAc (pH 5.3), 10 mM EDTA, 5 µg of tRNA (Roche 28473522), and 5 µg of glycogen and incubated at −20°C. The pellet was washed with 70% ethanol and resuspended in RNA loading buffer (formamide, 10 mM Tris-HCl at pH 8.0, 5 mM EDTA, 0.02% xylene cyanol). Samples were separated on 6% urea-polyacrylamide gels by electrophoresis and probed by Northern blotting, as detailed in the Supplemental Material. Phosphorimager scans were processed and quantitated with ImageJ software. The indicated pA tail lengths were approximated from DNA size markers.
Biotinylation and purification of 4tU-labeled RNA
Biotinylation and purification of newly synthesized RNA were based on the protocol by Barrass and Beggs (2019) with minor modifications detailed in the Supplemental Material.
[35S]-labeling of proteins
The [35S]-methionine labeling protocol was modified from Stutz et al. (1997) and Kallehauge et al. (2012) and is detailed in the Supplemental Material. Briefly, cells grown at 25°C in a synthetic complete minimal medium lacking methionine and containing 2% glucose (SC-met) were heat-shocked by adding an equal volume of medium preheated to 51°C and transferring to a 38°C water bath. For samples maintained at 25°C, an equal volume of 25°C medium was added. Rapamycin (final concentration 1 µg/mL) or DMSO (final 0.5%) was added to cell cultures before heat shock, as well as to the preheated medium, at a time point specified in the figure legends. Aliquot of cell suspension (0.6 OD units) was transferred at the indicated times to a new microfuge tube containing 50 µCi of [35S]-methionine (43.3 mCi/mL:1175 Ci/mmol; PerkinElmer NEG009C) to label the proteins in a total volume of 400 µL, and transferred to ice at the end of the labeling period. Cells were collected by centrifugation and washed twice with ice-cool water, and the cell pellets were stored at −20°C. The pellets were resuspended and boiled for 5 min in 30 µL of Laemmli buffer (60 mM Tris-Cl at pH 7.0, 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue), before running 15 µL of the supernatant on SDS-PAGE. Dried gel was exposed to a phosphorimager screen and quantitated using ImageJ software.
Expression and purification of recombinant CPF
Detailed methods for protein expression and purification are described in the Supplemental Material. Multibac transfer vectors containing CPF genes were used (Supplemental Table S4), as described previously (Kumar et al. 2021). The Pfs2 and Ref2 genes encoded a C-terminal Strep-II (SII) tag. Vectors carrying the polymerase module genes (Cft1, Yth1, Fip1, Pap1, and Pfs2-SII) or combined phosphatase and nuclease module genes (Pta1, Pti1, Ssu72, Swd2, Glc7, Ref2-SII, Ysh1, Cft2, and Mpe1) were transformed into chemically competent DH10EmbacY cells, and bacmids were isolated. Sf9 cells expressing either the polymerase module or the combined phosphatase and nuclease modules were lysed by sonication and protein complexes purified using affinity purification in batch (Strep-Tactic Superflow resin, IBA Lifesciences) followed by anion exchange chromatography (Mono Q 5/50 GL, Cytiva). Finally, the recombinant CPF complex was reconstituted by mixing the purified polymerase module and combined phosphatase and nuclease modules at 1:1 molar ratio and applied to a Superose 6 increase 3.2/300 column (Cytiva). The peak fractions were used directly in the polyadenylation reactions. All purifications were performed at 4°C.
Expression and purification of CF IA and Nab2p
Genes encoding CF IA subunits (Rna14, Pcf11, Rna15, and Clp1) were cloned into Multibac transfer vectors. The CF IA complex was purified by Strep-Tactin affinity followed by a HiTrap heparin HP column (Cytiva). The peak fractions were further purified by size exclusion chromatography, analyzed by SDS-PAGE, and flash-frozen in liquid nitrogen for storage at −80°C. Full-length Nab2p was cloned into a pACEBac1 expression vector and was purified by Strep-Tactin affinity followed by a 5-mL RESOURCE 15S column (Cytiva). The peak fractions were applied to a Superdex 200 increase 10/300 column (Cytiva), analyzed by SDS-PAGE, concentrated to 1 mg/mL, and flash-frozen in liquid nitrogen for storage at −80°C.
Expression and purification of CF IB and Pab1p
CF IB was expressed and purified as described previously (Casañal et al. 2017). A DNA sequence encoding full-length Pab1p from S. cerevisiae was codon-optimized for E. coli (GenScript) and cloned into a pGEX-6P-1 vector containing an N-terminal GST-tag with an HRV-3C protease cleavage site. Protein expression in BL21 DE3 was induced using 0.5 mM IPGT, cells harvested by centrifugation, and lysed by sonication. The clear supernatant was applied to a 5-mL GSTrap 4B column (Cytiva) followed by an overnight incubation at 4°C with a 1:50 molar ratio (protein:enzyme) of HRV-3C protease to remove the GST-tag. The peak fractions were pooled and applied to a Superdex 200 increase 10/300 column (Cytiva), analysed by SDS-PAGE, concentrated to 5 mg/mL, and flash-frozen in liquid nitrogen for storage at −80°C.
In vitro polyadenylation assays
Assays were performed at 30°C in a reaction buffer containing 5 mM HEPES (pH 7.9), 150 mM KAc, 2 mM MgAc, 0.05 mM EDTA supplemented with RiboLock RNase inhibitor (Thermo Scientific), and 3 mM DTT. Purified CPF was added to a final concentration of 50 nM in the reaction. CF IA was diluted to 15 µM in 20 mM HEPES (pH 8.0), 250 mM NaCl, and 0.5 mM TCEP and added to a final concentration of 450 nM. CF IB, Nab2p, and Pab1p were diluted in 50 mM HEPES (pH 7.9), 150 mM KCl, 0.5 mM MgAc, 1 mM TCEP and added to their final concentrations. CF IB concentration was 450 nM throughout the experiments, whereas Nab2p and Pab1p were added to a final concentration of 60 nM–1 µM as specified in the figures. RNA substrates were used at a final concentration of 100 nM. RNA sequences are listed in Supplemental Table S1. Reactions were initiated by addition of ATP to a final concentration of 2 mM.
Nanopore direct RNA sequencing
Two independent RNA samples were sequenced for each experimental condition, except for 25°C samples, which were sequenced only once. One 4tU RNA sample for each experimental condition was sequenced independently two or three times. Polyadenylated RNA (pA+ RNA) was purified from 35 µg of total RNA using the Dynabeads oligo dT(25) kit (Invitrogen 61005) following the manufacturer's protocol. Yeast pA+, or 4tU RNA purified as specified in the Supplemental Material, was mixed with human or murine cap-enriched mRNA, and sequencing libraries were prepared from 500 ng of this RNA mix (of which 50–200 ng consisted of yeast pA+ or 4tU RNA) with the direct RNA sequencing kit (Oxford Nanopore Technologies SQK-RNA002) according to the manufacturer's protocol. RNA was sequenced using the FLO-MIN106 flow cells and the MinION Mk1B device (Oxford Nanopore Technologies). Base-calling was performed with the software Guppy (Oxford Nanopore Technologies). Raw sequencing data (fast5 files) were deposited at ENA. All sequencing runs with their ENA accession numbers are listed in Supplemental Table S5. DRS libraries for samples MEX67-AA pPgal-empty at 25°C and at 38°C with or without rapamycin described by Tudek et al. (2021) were reanalyzed here.
Bioinformatic analyses
Yeast-originating DRS reads were separated from the other samples by mapping to the respective reference transcriptomes using Minimap2 (Li 2018), filtering out reads that were unmapped to the yeast reference, and mapping the remaining reads with Minimap 2.17 to a custom annotation of yeast transcriptome reference (Tudek et al. 2021) described in the Supplemental Material. The pA tail lengths for each read were estimated using the Nanopolish 0.13.2 polya function (Workman et al. 2019). pA tail length distribution graphs were generated using the ggplot2 package in R (Wickham 2016). hsRNAs were identified as transcripts with at least threefold change in expression at “38°C; no rapamycin; empty” sample compared with “25°C; empty” sample, mean expression >50 reads, and Benjamini–Hochberg adjusted P-value < 0.05, using binomial test from EdgeR (Robinson et al. 2010). This set of transcripts is listed in Supplemental Table S2. Full details of bioinformatic analyses including the procedure of fitting the pA tail lengths to bimodal distributions are described in the Supplemental Material. Processed data containing pA length estimates for each read, the custom code, and the yeast transcriptome reference are available at GitHub (https://github.com/THJlab/Turtola_etal).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Karsten Weis for providing the original pab1ΔRRM4 yeast strain, Maurice Swanson for the Nab2p antibody, and Terence Tang for pGEX6P-Pab1. David Barrass is acknowledged for providing guidance and protocols for 4tU RNA purification. Thomas Beuchert Kallehauge is acknowledged for design and preparation of the FISH probes. M.T. was supported by a Federation of European Biochemical Societies long-term fellowship and an EMBO long-term fellowship (ALTF 328-2019). M.C.M was supported by a Novo Nordisk Fonden grant (NNF19OC0054219), A.K. was supported by Gates Cambridge, and A.C. was supported by an EMBO long-term fellowship (ALTF66-2015) cofunded by the European Commission (LTFCOFUND2013 and GA-2013-609409) through Marie Curie Actions. S.M. was supported by a Polish National Science Centre grant (no. 2020/38/E/NZ2/00372). Funding for this work in T.H.J.’s laboratory was granted from Independent Research Fund Denmark. Work in A.D.’s laboratory was supported by a TEAM/2016-1/3 Foundation for Polish Science grant (to A.D.). A.D. is also a recipient of the European Research Area Chairs position funded by the European Union (agreement no. 810425). Work in L.A.P.’s laboratory was supported by the European Union's Horizon 2020 research and innovation program (ERC Consolidator grant agreement 725685) and the Medical Research Council (MRC) as part of United Kingdom Research and Innovation (MRC file reference no. MC_U105192715).
Author contributions: M.T., M.S., A.C., A.K., L.A.P., and T.H.J. conceived the study. M.T., M.C.M., A.K., M.S., and A.C. performed the methodology. P.S.K. and M.S. acquired and used the software. M.T., P.S.K., and M.S. performed the formal anaylsis. M.T., M.C.M., A.K., A.T., S.M., and A.C. performed the investigation. A.D. acquired the resources. P.S.K. and M.S. curated the data. M.T. and M.C.M. wrote the original manuscript draft. T.H.J., M.S., L.A.P., A.C., A.K., P.S.K., A.T., and A.D. reviewed and edited the manuscript. M.T., M.C.M., A.C., and M.S. visualized the study. L.A.P. and T.H.J. supervised the study. M.T. and A.C. were the project administrators. A.D., L.A.P., and T.H.J. acquired the funding.
Footnotes
-
↵Present addresses: 6Department of Molecular, Cellular, and Developmental Biology, Yale University, New Haven, CT 06511, USA; 7Fondazione Human Technopole, 20157 Milano, Italy.
-
Supplemental material is available for this article.
-
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348634.121.
-
Freely available online through the Genes & Development Open Access option.
- Received May 4, 2021.
- Accepted July 15, 2021.
This article, published in Genes & Development, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.









