Recurrent Horizontal Transfer of Bacterial Toxin Genes to Eukaryotes
- 1Department for Molecular Evolution and Development, Center for Organismal Systems Biology, Faculty of Life Sciences, University of Vienna, Vienna, Austria
- 2Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw, Poland
- 3Institute of Experimental Plant Biology, University of Warsaw, Warsaw, Poland
- ↵*Corresponding author:
E-mail: ulrich.technau{at}univie.ac.at; yehu.moran{at}univie.ac.at.
Abstract
In this work, we report likely recurrent horizontal (lateral) gene transfer events of genes encoding pore-forming toxins of the aerolysin family between species belonging to different kingdoms of life. Clustering based on pairwise similarity and phylogenetic analysis revealed several distinct aerolysin sequence groups, each containing proteins from multiple kingdoms of life. These results strongly support at least six independent transfer events between distantly related phyla in the evolutionary history of one protein family and discount selective retention of ancestral genes as a plausible explanation for this patchy phylogenetic distribution. We discuss the possible roles of these proteins and show evidence for a convergent new function in two extant species. We hypothesize that certain gene families are more likely to be maintained following horizontal gene transfer from commensal or pathogenic organism to its host if they 1) can function alone; and 2) are immediately beneficial for the ecology of the organism, as in the case of pore-forming toxins which can be utilized in multicellular organisms for defense and predation.
Key words
Introduction
Horizontal (lateral) gene transfer (HGT) between species from different kingdoms of life is considered a rare event, especially when “higher” organisms such as animals are involved (Keeling and Palmer 2008; Keeling 2009; Dunning Hotopp 2011). Still, the number of described cases of HGTs between kingdoms is consistently rising, with several striking examples demonstrated in recent years (e.g., Gladyshev et al. 2008; Moran and Jarvik 2010). Whereas the high abundance of HGT in prokaryotes allows study of trends in transfer and maintenance of specific gene families, the sporadic nature of HGT in multicellular eukaryotes makes similar analyses difficult. As rare as HGTs between kingdoms are, even rarer are cases of recurring horizontal transfers of genes from the same family between kingdoms. A notable exception is the independent transfers of cellulase genes from unicellular organisms to nematodes (Mayer et al. 2011).
Here, we report the recurrent HGT of members of the aerolysin gene family between various kingdoms of life. Aerolysin is a pore-forming toxin (PFT) from the pathogenic bacterium Aeromonas hydrophyla (Abrami et al. 2000). Its pore-forming protein domain is found in a family of bacterial toxins from various animal and human pathogens belonging to the Firmicute and Gammaproteobacteria phyla. Toxins of this family oligomerize into a β-barrel structure that disrupts host cell membranes, leading to lysis of the cell. Notably, several aerolysin homologues have been shown to be involved directly in pathogenicity (Kennedy et al. 2009; Popoff and Bouvet 2009). According to domain architecture and protein structure analyses, aerolysin-like proteins are present in a diverse group of organisms from all kingdoms of life: bacteria, archaea, fungi, animals, and plants (Mancheno et al. 2010; Szczesny et al. 2011). However, the vast majority of those proteins have low sequence similarity to aerolysin (<20% similarity), raising difficulties in resolution of their phylogeny by traditional methods.
Since aerolysin domains were detected in proteins from all kingdoms of life, it could be hypothesized that they all derive from an ancestral protein that was present in one of the early bacterial or archaeal lineages (and then spread by HGT), if not in the last universal common ancestor (LUCA) of all living cells. However, to our surprise, cluster mapping and phylogenetic analyses of aerolysin domains show that aerolysin proteins from species that are very distantly related are often significantly more similar to each other, than they are to proteins from more closely related species. This observation raises the possibility that HGT has played a significant role in the evolution of aerolysins. Moreover, their distribution suggests that multiple independent transfer events occurred in the evolution of the aerolysin protein family.
Materials and Methods
Identification of Aerolysin Homologues and Cluster Mapping
To obtain the list of sequences belonging to the aerolysin superfamily, we made iterative searches on nonredundant database (nr) from NCBI. We used sequences containing an aerolysin domain (from the alignment in Szczesny et al. (2011) as starting points for three rounds of searches with the Jackhmmer program from the HMMER3 package (http://hmmer.janelia.org). We used an e-value threshold of 0.001 at the protein domain level in all three iterations. The threshold was chosen based on the manual assessment of alignments done in the previous study (Szczesny et al. 2011). From the resulting high scoring pairs (HSPs), we collected those long enough to form a minimal aerolysin structural core (minimal length of 75 residues). This resulted in a total of 545 sequences that were compared all versus all with BLASTP (Altschul et al. 1990) and clustered using the CLANS software (Frickey and Lupas 2004). The final visualization was obtained with an e-value threshold of 0.01 applied in the CLANS program.
Phylogenetic Analysis
Sequences were aligned using MAFFT and low-quality alignment regions were removed by TrimAl (Katoh et al. 2005; Capella-Gutierrez et al. 2009). ProtTest was used to find the most suitable model for phylogeny reconstruction of aerolysins (Abascal et al. 2005). The maximum-likelihood (ML) phylogenetic tree was constructed using PhyML with the WAG Model (+F +G), which got the highest score in the ProtTest analysis (Guindon et al. 2010). Support values were calculated using 100 bootstrap replicates. A Bayesian tree was constructed using MrBayes version 3.1.2 with the WAG model. The run was performed for 5,000,000 generations and every 100th generation was sampled. We estimated that the Bayesian analysis reached convergence when the potential scale reduction factor reached 1.0. We tested phylogenetic hypotheses using the approximately unbiased (AU) test (Shimodaira 2002). For each tested tree, we calculated site-likelihoods using Tree-Puzzle (Schmidt et al. 2002) and performed the AU test using CONSEL (Shimodaira and Hasegawa 2001) with default scaling and replicate values.
To produce the species tree used for gene loss counting, we obtained a list of all currently sequenced genomes from NCBI Genome (http://www.ncbi.nlm.nih.gov/genome) and visualized the corresponding NCBI Taxonomy species tree (Federhen 2012) using iTol (Letunic and Bork 2011). The minimal number of losses was counted by collapsing all sister clades except those harboring the gene, and placing a loss event on the ancestral branch of each clade.
In Situ Hybridization
Total RNA was extracted from 12-days old Nematostella vectensis primary polyps using Trizol reagent (Invitrogen, USA). It served as a template for the synthesis of complementary DNA (cDNA) using the SuperScript III reverse transcriptase (Invitrogen). The primers 5'-TTCGCTGGGTGTCCCGTACTGCTG-3' and 5'-GTTTGTGTTAGTATCGGTGGTCGT-3' were used in polymerase chain reaction in order to amplify an Nv-Lysin1b fragment from the cDNA. The 1,086-bp DNA product was cloned into the pGEM-T Easy vector (Promega, USA). A digoxigenin (DIG) labeled probe was synthesized using the MegaScript kit (Ambion, USA) and a DIG RNA labeling mix (Roche, Germany). In situ hybridization (ISH) was carried out as previously described for Nematostella (Genikhovich and Technau 2009). For all ISH experiments, N. vectensis larvae were fixed at 24–144 hpf in ice-cold 3.7% formaldehyde in one-third seawater with 0.2% glutaraldehyde for 90 s and then in 3.7% formaldehyde in one-third seawater with no glutaraldehyde for additional 60 min. Probe generation of hydralysin and ISH experiments in Hydra viridissima were conducted as previously described (Sher et al. 2008).
Results
Bacterial and Eukaryotic Aerolysin Homologues Cluster Together in Sequence Comparisons
More than 300 members of the aerolysin superfamily were collected based on protein domain content and clustered based on basic local alignment search tool (BLAST) pairwise identity using the CLANS software (see Materials and Methods). We defined distinct clans according to an e-value threshold of 0.01. Two central and well-supported clans contained aerolysin homologues from species of more than one kingdom of life (fig. 1). To further examine this result, we performed a phylogenetic analysis on the members of the two clans as well as adjacent sequences that were members of the aerolysin domain seed alignment in PFAM (PF01117). We focused on those clans, because the central region is more reliable than the outer sparser parts of the cluster graph, thanks to a larger number of independent supporting pairwise identities, allowing us to construct an informative multiple sequence alignment. ML and Bayesian analyses provided highly similar tree topologies (fig. 2). The phylogeny revealed multiple clusters that comprised sequences from very different organisms. Cluster 1 containing putative aerolysin homologues from the sea anemone N. vectensis were positioned as a sister group to PFTs of the pathogenic bacterial groups Aeromonas, Vibrio, Clostridium, and other Firmicutes and Gammaproteobacteria. The cytolytic toxin enterolobin from the plant Enterolobium contortisiliquum also fell within this bacterial group (Sousa et al. 1994). In cluster 2, aerolysin homologues from the fungal plant parasite Coprinopsis cinerea and the hemolytic lectins from the pathogenic fungus Laetiporus sulphureus (Tateno and Goldstein 2003) clustered with proteins from several plants, including species of pivotal agricultural importance such as bread wheat (Triticum aestivum) and wine grape (Vitis vinifera) (Szczesny et al. 2011). Cluster 3 contained multiple genes of the tick Ixodes scapularis encoding aerolysin homologues, that clustered with toxins from the bacterial pathogens Pseudomonas aeruginosa and Pectobacterium wasabiae. Cluster 4 contained hydralysins, PFTs from three species of the cnidarian genus Hydra that closely clustered with the cytolytic protein parasporin from Bacillus thuringiensis (Ito et al. 2004; Sher et al. 2005). Although cluster 1 is the combination of two clans and a single protein (enterolobin) from the clan map, these sequences were grouped together in the phylogenetic tree with relatively high support (bootstrap support of 85 and posterior probability of 1.0; fig. 2). Clusters 3 and 4 in the phylogenetic tree both derived from one cluster in the CLAN map (fig. 1). Cluster 2 was also retrieved by the CLAN map but is part of a bigger cluster which also contains aerolysin domains from many more groups such as archaea and lepidopterans (Szczesny et al. 2011). In general, the support values for nodes in cluster 2 were lower than for other nodes (fig. 2) and including all the diverse protein sequences of cluster 2 in the alignment resulted in much lower bootstrap support and posterior probability for that part of the tree (data not shown). For that reason, we only included the plant and fungi aerolysin homologues in the final tree. To assess the confidence in the maximum likelihood topology shown in figure 2, we compared it to an alternative topology where all eukaryotic sequences were constrained to be monophyletic, using the approximately AU test (Shimodaira 2002). The monophyletic tree was rejected at a high confidence level (delta log-likelihood 153.7, P value 8 × 10−5), in support of an evolutionary scenario involving HGT.
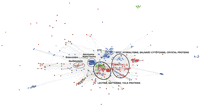
Cluster map of aerolysin homologues. Protein sequences corresponding to aerolysin domains were clustered based on their pairwise similarity using the CLANS program. The clans containing sequences from more than one kingdom of life are indicated by black circles. Clans containing members of the PFAM aerolysin seed alignment are indicated by dashed circles. Bacteria appear in blue, plants in green, fungi in brown, and animals in red. GenInfo Identifier (GI) numbers of proteins appear in supplementary table 1 (Supplementary Material online).
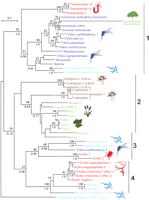
Phylogeny of aerolysin homologues from different kingdoms of life. A ML phylogenetic tree of aerolysin homologues from different organisms was constructed with the WAG model (+F, +G). The organisms that produce the aerolysins are indicated by genus names. In cases where several species of the same genus produce aerolysin-like proteins, the full species names are indicated. The tree is rooted with the epsilon-toxin from Clostridium perfringens. GI numbers of proteins appear in supplementary table 1 (Supplementary Material online.) Bootstrap support values above 50% are indicated above branches and Bayesian posterior probability values above 0.75 appear below branches. Gammaproteobacteria appear in dark blue, Firmicute bacteria in light blue, plants in green, fungi in brown, and animals in red. The corresponding cluster numbers from the clans analysis appear to the left of the tree.
So far, only two toxins containing aerolysin domains were studied thoroughly at the structure–function level by mutagenesis accompanied by binding and activity assays: aerolysin from A. hydrophyla and the Alpha-toxin from Clostridium septicum (Buckley et al. 1995; MacKenzie et al. 1999; Melton-Witt et al. 2006). Comparison of their sequences to those of the plant toxin enterolobin and to the putative aerolysin protein from the cnidarian N. vectensis revealed that 4 or 6 of 9, respectively, of the residues shown to have a functional role in the bacterial toxins are conserved in these two eukaryotic counterparts. Many of these residues are embedded in conserved motifs, suggesting that these sites still have a similar function. (fig. 3).

Conservation of functional residues in aerolysin eukaryotic homologues. Multiple sequence alignment of aerolysin domains of aerolysin from the bacterium Aeromonas hydrophyla (GenBank accession 1pre_A), enterolobin cytotoxin from the plant Enterolobium contortisiliquum (Swiss-Prot accession P81007), Clostridial α-toxin from the bacterium Clostridium septicum (GenBank accession Q8GI65) and an aerolysin homologue from the sea anemone Nematostella vectensis (NCBI reference XP_001634534). Conserved positions appear in black font on light gray background. Positions proved by mutagenesis to be involved in activity, are indicated by asterisk (MacKenzie et al. 1999; Kennedy et al. 2009). Two positions previously shown to be involved in oligomerization of aerolysin are indicated by the pound sign (Buckley et al. 1995). The position originally mutated appears in bold, conserved residues at these positions appear on black background, and conservative substitutions are in white font on dark gray background.
Aerolysin Homologues from Two Cnidarians Species Vastly Differ in Gene Structure and Expression Patterns
We decided to further study the aerolysin homologues of the two cnidarians Nematostella and Hydra as examples for proteins that belong to species of the same phylum, but whose aerolysin sequences cluster with proteins from very different phyletic groups (figs. 1 and 2). We first assayed their expression patterns by in situ hybridization. It is commonly observed that developmental regulators as well as regional and cell-type markers are often expressed in homologous manner in Hydra and Nematostella (e.g., Technau, 2001; Swalla 2006; David et al., 2008; Zenkert et al. 2011). If the aerolysin genes were inherited from a common cnidarian ancestor, the likelihood of a common expression pattern would be relatively high. In contrast, if the genes were acquired independently, the likelihood of a common expression pattern is minimal. While the expression of hydralysins in H. viridissima is widespread in digestive endodermal cells of the gastrovascular cavity (Sher et al. 2008), we found that the N. vectensis aerolysin homologue Nvlysin1b is expressed in distinct large gland cells in the ectoderm of the pharynx (fig. 4). Hence, Nematostella and Hydra aerolysin genes are expressed in nonhomologous tissues and cells, consistent with an independent acquisition of the genes. Next, we examined their gene structures based on gene models in the available genome sequences (Putnam et al. 2007; Chapman et al. 2010). Whereas the genes encoding the hydralysins of H. magnipapillata have no introns, those encoding the aerolysin homologues in N. vectensis carry five introns (supplementary fig. S1, Supplementary Material online).
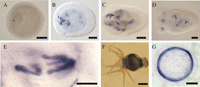
Expression patterns of aerolysin homologues in two cnidarian species. ISH was used in order to localize the expression of Nvlysin-1b in Nematostella vectensis and hydralysin-2 (Hln-2) in Hydra viridissima. In N. vectensis the expression was restricted to distinct large ectodermal cells in the pharynx (dark blue staining). Expression started in very few cells in 2-days old animals (A) and the domain expanded with age (B: 3-days old planula; C: 4-days old planula; D: 6-days old primary polyp). Nvlysin-1b is expressed in large cells (>40 μm), which probably are gland cells (unstained round spaces inside stained cells are vesicles, E). In H. viridissima, the aerolysin homologue Hln-2 is expressed in all endodermal cells in the gastrovascular cavity as shown by blue-purple staining in whole mount ISH (F) and a cross section of a stained animal (G). Scale bars are 50 μm in panels A–D, 40 μm in panel E, 500 μm in F, and 100 μm in G.
Hydralysins were shown to be secreted during feeding and are likely to be involved in disintegration of crustacean prey in H. viridissima (Sher et al. 2008). We hypothesize that the Nematostella homologues may play a similar role in disintegrating and killing paralyzed prey as they are produced in ectodermal gland cells of the pharynx where they directly come in contact with prey when swallowed (fig. 4).
Discussion
Cluster mapping and phylogenetic analysis of aerolysin proteins reveal that sequences from remotely related organisms are more closely related than expected, and cluster together in a way that is vastly different from the species phylogeny (figs. 1 and 2); aerolysin domains from species which belong to the same phylum or the same kingdom of life are located in distinct clusters. Notable examples are the two cnidarian aerolysin groups, each of which clusters with a distinct bacterial group, and the enterolobin toxin from the plant E. contortisiliquum, which is located within a bacterial aerolysin cluster and is clearly separated from the other plant aerolysin homologues (fig. 2). The example of vastly different expression patterns and gene structures of the aerolysin homologues in the two cnidarian groups fits with the idea that aerolysins in species of the same phylum can be of very different origins (fig. 4).
An extremely patchy phylogeny and an unusual clustering of sequences from different kingdoms of life can result from three different evolutionary scenarios: 1) convergent evolution, 2) massive gene loss in most lineages, and 3) recurrent HGT. Convergent evolution is a very unlikely explanation for the closely related aerolysin sequences from different phyla. Many of those proteins exhibit around 60% similarity despite being found in organisms as distant as a bacterium and an animal, while proteins from the same phylum (e.g., Cnidaria) may show only 20% similarity. Functional studies using mutagenesis identified a very small set of constrained amino acids (MacKenzie et al. 1999). This demonstrates that the sequence space in which we find aerolysin structure and function is not unusually constrained. Following this argument, it is highly unlikely that such protein similarity clusters appeared convergently in the course of evolution.
Inheritance of an ancestral gene accompanied by multiple gene losses is a popular explanation for patchy gene distributions, as gene losses are suggested to be much more common events than HGT as demonstrated in parasite genomes (Moran 2002; Koonin 2003; Technau et al. 2005; Keeling and Palmer 2008).
Indeed, in the case of the cnidarian Nematostella, we assume that the majority of expressed sequence tags initially found with best nonmetazoan hits could be explained by a combination of gene loss and divergence (Technau et al. 2005; Fredman and Technau, unpublished data). However, in the case of aerolysin, we detect several distinct clusters containing proteins derived from two distinct kingdoms of life. Thus, a scenario relying solely on differential gene loss would require not only the emergence of one ancestral aerolysin in LUCA, but that LUCA had multiple aerolysin copies which later diverged. This appears improbable, if estimates that LUCA was an organism with a limited gene content estimated at 500–1,000 genes are correct (Koonin 2003; Mushegian 2008). Moreover, an extraordinary amount of independent gene loss events in each of the lineages diverging from LUCA would be required to explain the phylogeny if no HGT is involved. Taking the clustering of the Ixodes and Pseudomonas proteins as an example, we estimate based on the currently available phylogenetic tree provided by NCBI taxonomy (Federhen 2012) pruned to represent organisms whose genome has been sequenced (1,150 prokaryotes and 366 eukaryotes at the time of writing), that a minimum of 76 losses would be required (52 in prokaryotes and 24 in eukaryotes, supplementary fig. S2, Supplementary Material online). Similar numbers would be required to explain each of the other clusters, for a total of several hundred independent losses. This is an underestimation as multiple losses in the same lineage cannot be accounted for. Moreover, the phylogenetic groups in which these proteins are present are well represented in public databases. With so many prokaryote and eukaryote genomes now available, lack of sequenced genomes cannot be held responsible for the patchy aerolysin distribution like in past reports (Keeling and Palmer 2008).
Thus, we consider recurrent HGT events to be the most likely explanation for the unusual aerolysin phylogeny. The fact that several aerolysin homologues from bacteria were found on phages and plasmids provides further support for an HGT scenario, especially within prokaryotes, and it is possible that these vectors also contribute to the mobilization of aerolysin genes between diverse lineages (Nakayama et al. 1999; Miyamoto et al. 2008; Gonzalez et al. 2011). If we consider only clusters with reliable phylogeny, then at least six HGT events between kingdoms of life have occurred in the evolution of the aerolysin family. To the best of our knowledge, this is the first report describing such magnitude of recurrent cross-kingdom HGT events within the same gene family. Recently, a family of DNA transposons named SPINs was proposed to be recurrently transferred between tetrapods in unprecedented numbers (Pace et al. 2008; Gilbert et al. 2012). Although the transfers of SPINs are limited to one vertebrate group, the recurrent transfers of these elements demonstrate HGT among “higher” eukaryotes may be more common than initially proposed.
It is apparent that none of the HGT events shaping the evolution of the aerolysin family are recent because all the eukaryotic aerolysin genes, with the exception of the hydralysins, contain introns and vary in gene structure from one another (supplementary fig. S1, Supplementary Material online). Even the intronless hydralysins that cluster with bacterial parasporins are the result of an HGT that occurred at least 60 Ma, as this is the estimated divergence time of the hydralysin producing species H. viridissima and H. vulgaris (Martinez et al. 2010). If the proteins are of bacterial origin, the age of the proposed HGT events in the aerolysin family is more than sufficient to eliminate traces of recent HGT, such as gene structure, synteny, and GC content. In general, these limitations make documented cases of ancient HGT very rare compared with recent HGTs.
Interestingly, two other PFT families exhibit a highly unusual patchy distribution across kingdoms and phyla; aegerolysins that are PFTs found in bacteria, fungi, and plants, and actinoporins found in cnidarians, fish, mosses, and other basal land plants. HGT was previously suggested for both classes but the supporting evidence was very limited (Berne et al. 2009; Hoang et al. 2009). If HGT events are rare, how could one explain the even rarer cases of reoccurring HGT events, exemplified here by the aerolysin family? What makes PFTs so unique that possibly three distinct types have repeatedly been transferred horizontally between different kingdoms of life? We suggest that certain gene families are more likely to be maintained after horizontal transfer if they can serve as “self contained units,” meaning that they can function alone and are not part of protein complexes or complicated pathways. A similar reasoning was put forward under the “Complexity hypothesis” to explain the much more common HGTs among bacteria (Jain et al. 1999). Although this hypothesis was recently challenged in a bacterial context (Gophna and Ofran 2011), it might still apply to cross-kingdom HGTs, since those introduce foreign proteins into a new and very different cellular environment. Another factor that would make a gene a good HGT candidate is the ability to immediately benefit and fit the ecology of the organism. Indeed PFTs meet both qualifications as they self-oligomerize and require no other proteins for their activity. As such, they could easily be adopted as functional units in animals, plants, and fungi. We suggest that cellulases which were transferred from various microbes to nematodes, may also meet these qualifications; Many nematode cellulases are “self contained units” that contain both carbohydrate binding and catalytic domains (Kyndt et al. 2008; Mayer et al. 2011), and the ability to hydrolyze cellulose or other complex carbohydrates is of immediate advantage for nematodes feeding on plants or on microorganisms (Mayer et al. 2011). Like in the case of many HGT events between prokaryotes, it can be suggested that many genes transferred between kingdoms are preadaptive traits, with a neutral or nearly neutral value, that may become advantageous for the host only upon subsequent environmental changes (Gogarten and Townsend 2005). However, for the case of recurrent transfer and retention in a diverse set of species of proteins belonging to one specific protein family, a model that allows for immediate positive selection seems more likely.
While the most obvious PFT functions are in defense and predation, many of the PFTs and their homologues are expressed in nonvenomous, nontoxic, and nonpathogenic species, raising questions regarding their role in those organisms (Hoang et al. 2009; Szczesny et al. 2011). Nevertheless, there are good indications that PFTs may also serve as pore-formers in a nonvenomous context, for example in digestion. A clear example is that of hydralysins, which serve in hydras for prey disintegration after predation (Sher et al. 2008). The same role might be suggested for Nematostella aerolysins. In the tick, aerolysin homologues were found in salivary gland libraries. Because ticks feed exclusively on blood, we hypothesize that these PFTs likely serve in ticks to lyse devoured blood cells, a role previously suggested for PFTs in hematophagous insects (Amino et al. 2002). Another clear example for a nonvenomous role of PFTs is that of the moss actinoporin, which can lyse mammalian cells but serves as an intrinsic regulator of water stress (Hoang et al. 2009). Thus, the set of possible biological roles of PFTs and their homologues is probably wider than the one suggested for classic toxins, making them even more attractive candidates for adoption and maintenance following HGT events.
Supplementary Material
Supplementary figures S1–S2 and table 1 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank Daniel Sher (University of Haifa) for sharing data and for critically reading the manuscript. They also thank Thomas Rattei and Matthias Horn (University of Vienna) for helpful discussions. This work was supported by an Initiativ Kolleg “Symbiotic Interactions” of the University of Vienna and grants of the Austrian National Science Foundation Fonds zur Förderung der wissenschaftlichen Forschung (FWF) (P22618-B17) to U.T. Y.M. was supported by a European Molecular Biology Organization (EMBO) long-term fellowship ALTF 1096-2009.
- © The Author(s) 2012. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0), which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.









